

Berrar D. et al. - Practical Approach to Microarray Data Analysis
.pdf194 |
Chapter 10 |
where  if the gene j is selected as the “winner” in method k;
if the gene j is selected as the “winner” in method k;  if the gene j is not selected as the “winner” in method k. If gene j is selected by all methods then
if the gene j is not selected as the “winner” in method k. If gene j is selected by all methods then 
The and  can be determined by other methods, too. For example, we may assign
can be determined by other methods, too. For example, we may assign  for all K methods used in variable selection stage if we believe they perform equally well. We may also modify
for all K methods used in variable selection stage if we believe they perform equally well. We may also modify 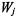 as
as
In this case, if gene j is selected by all methods and the Info  then
then 
The WFCCM also reduces the data dimension from to We can certainly view WFCCM as the “overall score” of each tissue sample, which combines all information of all important genes from several statistical methods.
3.3Leave-One-Out Cross-Validated Class Prediction Model
The misclassification rate can be assessed using leave-one-out crossvalidated (LOOCV) class prediction method. Cross-validation is a method for estimating generalization error based on resampling. LOOCV is one specific type of cross-validation. The LOOCV is processed in four steps in the Vanderbilt lung cancer SPORE study. First, apply the WFCCM to calculate the single compound covariate for each tissue sample based on the significant genes. Second, one tissue sample is selected and removed from the dataset, and the distance between two tissue classes for the remaining tissue samples is calculated. Third, the removed tissue sample is classified based on the closeness of the distance of two tissue classes, e.g., k-nearest neighbor approach, which k=2, or using the midpoint of the means of the WFCCM for the two classes as the threshold. Fourth, repeat step 2 and 3 for each tissue sample. To determine whether the accuracy for predicting membership of tissue samples into the given classes (as measured by the number of correct classifications) is better than the accuracy that may be attained for predicting membership into random grouping of the tissue samples, we may create 2,000-5,000 random data sets by permuting class labels among the tissue samples. Cross-validated class prediction is performed on the resulting data sets and the percentage of permutations that results in as few or fewer misclassifications as for the original labeling of samples can be reported. If less than 5% of the permutations result in as few or fewer misclassifications, the accuracy of the prediction of the given classes is considered significant. Therefore, this rate may be considered as the “p-value” for the class prediction model. We recently have succeeded in

10. Weighted Flexible Compound Covariate Method for Classifying |
195 |
Microarray Data |
|
applying the WFCCM class prediction analysis method to the preliminary data generated by the Vanderbilt lung cancer SPORE study.
The perfect WFCCM class-prediction models based on 54, 62, 77 and 27 differentially expressed genes were found to classify tumor tissue samples vs. normal tissue samples, primary lung cancer tissue samples vs. nonprimary lung cancer tissue samples, non-small cell lung cancer (NCLS) tissue samples vs. normal tissue samples as well as adenocarcinoma tissue samples vs. squamous cell carcinoma tissue samples. Table 10.1 shows the results from the Vanderbilt lung cancer study. WGA, SAM, Info-Score, and permutation t-test were applied in the analysis. The cut-off points were 3.0, 3.7 and p < 0.0001 for WGA, SAM, and Permutation t-test respectively. We selected  for all methods, and
for all methods, and 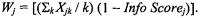
We also applied the WFCCM to a set of blinded/test samples. Table 10.2 shows the results of the analyses. In general, the model performed reasonably well (the average correct prediction rate in the blinded/test data set was 93%) except for predicting large cell vs. non-large cell tissues. Because there were only two genes reached the selection criteria in WFCCM for comparing large cell tissues with non-large cell tissues, this result was not a surprise.

196 |
Chapter 10 |
Figure 10.2 shows the results from the agglomerative hierarchical clustering algorithm for clustering adenocarcinoma and squamous cell carcinomas. The average linkage algorithm was applied to calculate the distance between the clusters. All the adenocarcinoma tissues clustered together, so did squamous cell tissues. The results looked very promising but we might only use these results to reconfirm the genes performed differently between two classes. Having a perfect or near perfect cluster result was expected if we applied the cluster analysis after we selected the genes that performed differently using any of the supervised methods. It is important to know that we could not apply these results in any class discovery conclusion!

10. Weighted Flexible Compound Covariate Method for Classifying |
197 |
Microarray Data |
|
4.CLASSIFICATION TREE METHODS
We have reviewed several class comparison methods and class prediction methods separately in this chapter. In this section, we would like to discuss some basic concepts of the classification tree methods since constructing classification trees may be seen as a type of variable selection while at the same time a predictive model is developed. Figure 10.3 shows the diagram illustrating a (simplified) decision tree for lung cancer risk evaluation. Classification trees are the structures that rigorously define and link choices and possible outcomes (Reggia, 1985).
The ideal situation for using classification tree methods is when there is a partition of the space  that will correctly classify all observations, and the task is to find a tree to describe it succinctly. In some cases the distributions of the classes over
that will correctly classify all observations, and the task is to find a tree to describe it succinctly. In some cases the distributions of the classes over 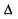 overlap, so there is no partition that completely describes the classes. Then, for each cell of the partition, there will be a probability distribution over the classes, and the Bayes decision rule will choose the class with highest probability. The training set idea described above can also apply to classification tree methods. The misclassification rate can be calculated by the proportion of the training set that is misclassified, and the generalization ability is estimated. With “noisy” data, such as microarray data, it is quite possible to construct a tree which fits the training set well, but which has adapted too well to features of that particular
overlap, so there is no partition that completely describes the classes. Then, for each cell of the partition, there will be a probability distribution over the classes, and the Bayes decision rule will choose the class with highest probability. The training set idea described above can also apply to classification tree methods. The misclassification rate can be calculated by the proportion of the training set that is misclassified, and the generalization ability is estimated. With “noisy” data, such as microarray data, it is quite possible to construct a tree which fits the training set well, but which has adapted too well to features of that particular

198 |
Chapter 10 |
subset of |
In other words, it can be too elaborate and over-fit the training |
data set. Overfitting is always a problem in any classifier.
Ji and Kim (unpublished manuscript) successfully applied the classification tree methods to classify toxic chemicals based on gene expression levels. The approach chooses the predictive genes as well as determines the classification rule. Three classification tree methods investigated are described below.
CART (Breiman et al., 1984) which stands for “Classification and Regression Trees” is biased when there are categorical predictors and when there are many missing values. With gene expression data, neither is the case, so CART gives a good fit. However, since it uses exhaustive search for variable selection, it often causes model overfitting. The CART algorithm is likely to give high accuracy in classification, but the genes selected may not be the most predictive ones.
QUEST (Loh and Shih, 1997) which stands for “Quick, Unbiased, Efficient Statistical Trees” is a program for tree-structured classification. The main strengths of QUEST are unbiased variable selection and fast computational speed. Also it is sensitive in identifying predictive genes since it uses statistical tests instead of exhaustive search. A common criticism of classification trees is that the construction of a classification tree can be extremely time-consuming. In addition, QUEST has options to perform CART-style exhaustive search and cost-complexity cross-validation pruning.
CRUISE (Kim and Loh, 2001), which stands for “Classification Rule with Unbiased Interaction Selection and Estimation”, consists of several algorithms for the construction of classification trees. It provides the features of unbiased variable selection via bootstrap calibration, multi-way splits for more than two classes, missing value treatment by global imputation or by node-wise imputation, and choice of tree pruning by cross-validation or by a test sample. CRUISE differs from QUEST and CART in that it allows multiway splits, which is natural when there are multiple classes.
Different classification tree methods may identify different sets of genes that have the same or similar misclassification rates. The concept of the WFCCM may be applied in this situation for combining all the possible “winners” genes from different classification tree methods.
5.CONCLUSION
The statistical class comparison and class prediction analyses for the microarray data may focus on the following steps: (1) Selecting the important gene patterns that perform differently among the study groups, (2) Using the class prediction model based upon the Weighted Flexible Compound Covariate Method (WFCCM), classification tree methods, or
10. Weighted Flexible Compound Covariate Method for Classifying |
199 |
Microarray Data |
|
other methods to verify if the genes selected in step one have the statistical significant prediction power on the training samples, (3) Applying the prediction model generated from step two to a set of test samples for examining the prediction power on the test samples, and (4) Employing the agglomerative hierarchical clustering algorithm to investigate the pattern among the significant discriminator genes as well as the biologic status.
The selection of important gene patterns may be based on different methods, such as Significance Analysis of Microarrays (SAM), Weighted Gene Analysis (WGA), and the permutation t-test. The cutoff points may be determined based on the significance as well as the prediction power of each method. The genes will be on the final list if they are selected by at least one of the methods.
The weighted flexible compound covariate method may be employed for the class-prediction model based on the selected genes. This method was designed to combine the most significant genes associated with the biologic status from each analysis method. The WFCCM is an extension of the compound covariate method, which allows considering more than one statistical analysis method into the compound covariate. The class prediction model can be applied to determine whether the patterns of gene expression could be used to classify tissue samples into two or more classes according to the chosen parameter, e.g., normal tissue vs. tumor tissue. We reviewed the leave-one-out cross-validated class prediction method based on the WFCCM to estimate the misclassification rate. The random permutation method may be applied to determine whether the accuracy of prediction is significant.
Applying the results of WFCCM from the training data set to the test samples is highly recommended. The test sample can be classified based on the closeness of the distance of two tissue classes, which is determined using the WFCCM in the training data set.
The classification tree methods have the features of variable selection and class prediction. Applying the concept of WFCCM for combining different “winners’ genes from different tree classification methods may be necessary.
ACKNOWLEDGEMENTS
This work was supported in part by Lung Cancer SPORE (Special Program of Research Excellence) (P50 CA90949) and Cancer Center Support Grant (CCSG) (P30 CA68485) for Shyr and by CCSG (P30 CA14520) for Kim. The authors thank Dr. David Carbone, PI of Vanderbilt Lung Cancer SPORE, for permission to use the study data for the illustration. The authors also thank Dr. Noboru Yamagata for his valuable suggestions.
200 |
Chapter 10 |
REFERENCES
Ben-Dor A., Friedman N., Yakhini Z. (2000). Scoring genes for relevance. Tech Report AGL- 2000-13, Agilent Labs, Agilent Technologies.
Bittner M., Chen Y. (2001). Statistical methods; Identification of differentially expressed genes by weighted gene analysis.
Available at http://www.nejm.org/general/content/supplemental/hedenfalk/index.html.
Breiman L., Friedman J.H., Olshen R.A., Stone C.J. (1984), Classification and Regression Trees. Wadsworth.
Ge N., Huang F., Shaw P., Wu C.F.J. PIDEX: A statistical approach for screening differentially expressed genes using microarray analysis (unpublished manuscript).
Hedenfalk I., Duggan D., Chen Y., Radmacher M., Bittner M., Simon R., Meltzer P., Gusterson B., Esteller M., Kallioniemi O.P., Wilfond B., Borg A., Trent J. (2001). Geneexpression profiles in hereditary breast cancer. N Engl J Med 344(8): 539-548.
Ji Y., Kim K. Identification of gene sets from cDNA microarrays using classification trees (unpublished manuscript).
Kim H., Loh W.-Y. (2001). Classification trees with unbiased multiway splits. J. American Statistical Association 96:589-604.
Landis S.H., Murray T., Bolden S., Wingo P.A. (1999). Cancer Statistics, 1999. CA Cancer J Clin 49(l):8-31.
Loh W.-Y., and Shih, Y.-S. (1997). Split selection methods for classification trees. Statistica Sinica 7:815-840.
Radmacher M.D., Simon R. Statistical methods: Generating gene lists with permutation F and t Tests.
Available at http://www.nejm.org/general/content/supplemental/hedenfalk/index.html.
Reggia J.A., Tuhrim S. (1985). An overview of methods for computer assisted medical decision making, in Computer-Assisted Medical Decision Making, Vol. 1, J.A. Reggia & S.Tuhrim, eds. Springer-Verlag, New York.
Shyr Y. (2002). Analysis and interpretation of array data in human lung cancer using statistical class-prediction model. AACR meeting April, 2002; San Francisco, CA: Program/Proceeding Supplement, 41-42.
Tukey J.W. (1993). Tightening the clinical trial. Control Clin Trials 14(4): 266-285.
Tusher V.G., Tibshirani R., Chu G. (2001). Significance analysis of microarays applied to the ionizing radiation response, Proc Natl Acad Sci USA 98(9): 5116-5121.
Wolfinger R.D., Gibson G., Wolfinger E.D. (2001). Assessing gene significance from cDNA microarray expression data via mixed models. J Comput Biol 8(6): 625-37.
Chapter 11
CLASSIFICATION OF EXPRESSION PATTERNS USING ARTIFICIAL NEURAL NETWORKS
Markus Ringnér1,2, Patrik Edén2, Peter Johansson2
1 Cancer Genetics Branch, National Human Research Institute, National Institutes of Health, Bethesda, Maryland 20892, USA
2 Complex Systems Division, Department of Theoretical Physics, Lund University, Lund, Sweden, e-mail: {markus,patrik,peterjg}@thep.lu.se
1.INTRODUCTION
Artificial neural networks in the form of feed-forward networks (ANNs) have emerged as a practical technology for classification with applications in many fields. ANNs have in particular been used in applications for many biological systems (see (Almeida, 2002) for a review). For a general introduction to ANNs and their applications we refer the reader to the book by Bishop (Bishop, 1995). In this chapter we will show how ANNs can be used for classification of microarray experiments. To this aim, we will go through in detail a classification procedure shown to give good results and use the publicly available data set of small round blue-cell tumors (SRBCTs) (Khan et al., 2001) as an example1. The ANNs described in this chapter perform supervised learning, which means that the ANNs are calibrated to classify samples using a training set for which the desired target value of each sample is known and specified. The aim of this learning procedure is to find a mapping from input patterns to targets, in this case a mapping from gene expression patterns to classes or continuous values associated with samples. Unsupervised learning is another form of learning that does not require the specification of target data. In unsupervised learning the goal may instead be to discover clusters or other structures in the data. Unsupervised methods have been used extensively to analyze array data and
1 The data is available at http://www.nhgri.nih.gov/DIR/Microarray/Supplement/.
202 Chapter 11
are described in other chapters. The main reasons for choosing a supervised method are to obtain a classifier or predictor, and to extract the genes important for the classification. Here we will exemplify this by describing an ANN based classification of expression profiles of SRBCT samples into four distinct diagnostic categories: neuroblastoma (NB), rhabdomyosarcoma (RMS), Burkitt’s lymphoma (BL) and Ewing’s sarcoma (EWS). This data set consists of 63 training samples each belonging to one of the four categories and 25 test samples. There are many other supervised methods (discussed in other chapters) that have been used to classify array data, spanning from simple linear single gene discriminators to machine learning approaches similar to ANNs, in particular support vector machines (SVMs). A major advantage of using a machine learning approach such as ANNs is that one gains flexibility. Using an ANN framework, it is for example straightforward to modify the number of classes, or to construct both linear and non-linear classifiers. Sometimes this flexibility is gained at the expense of an intuitive understanding of how and why classification of a particular problem gives good results. We hope this chapter will provide the reader with an understanding of ANNs, such that some transparency is regained and the reader will feel confident in using a machine learning approach to analyze array data.
We begin with a discussion on how to reduce high-dimensional array data to make the search for good ANNs more effcient. This is followed by section 3 on ANNs. Section 3 is split into 5 subsections as follows. We describe how to design an ANN for classification, how to train the ANN to give small classification errors, how a cross-validation scheme can be used to obtain classifiers with good predictive ability, how random permutation tests can be used to assess the significance of classification results, and finally how one can extract genes important for the classification from ANNs. This chapter ends with a short section on implementation followed by a summary.
2.DIMENSIONAL REDUCTION
For each sample in a typical study, the expression levels of several thousand genes are measured. Thus, each sample can be considered a point in ”genespace”, where the dimensionality is very high. The number of considered samples, N, is usually of the order of 100, which is much smaller than the number of genes. As discussed below, an ANN using more inputs than available samples tend to become a poor predictor, and in microarray analyses it is therefore important to reduce the dimensionality before starting to train the ANN. Dimensional reduction and input selection in connection with supervised learning have attracted a lot of interest (Nguyen and Rocke, 2002).
11. Classification of Expression Patterns Using Artificial Neural |
203 |
Networks |
|
The simplest way to reduce dimensionality is to select a few genes, expected to be relevant, and ignore the others. However, if the selection procedure involves tools less flexible than ANNs, the full potential of the ANN approach is lost. It is therefore preferable to combine the genes into a smaller set of components, and then chose among these for the ANN inputs.
For classification, we only need the relative positions of the samples in gene-space. This makes it possible to significantly reduce the dimensionality of microarray data without any loss of information relevant for classification. Consider the simple case of only two samples. We can then define one component as a linear combination of genes, corresponding to the line in gene-space going through the two sample points. This single component then fully specifies the distance in gene-space between the two samples. In the case of three samples, we can define as components two different linear combinations of genes, which together define a plane in gene-space which is going through the three data points. These two components then fully specify the relative location of the samples. This generalizes to N samples, whose relative locations in gene-space can be fully specified in an N - 1 dimensional subspace. Thus, with N samples, all information relevant for classification can be contained in N - 1 components, which is significantly less than the number of genes.
Reducing a large set of components (in our case, the genes) into a smaller set, where each new component is a linear combination of the original ones, is called a linear projection. In connection with ANNs, principal component analysis, PCA, is a suitable form of linear projection. PCA is described in detail in Chapter 5. In brief, it ranks the components according to the amount of variance along them, and maximizes the variance in the first components. Thus, the first component is along the direction which maximizes the variance of data points. The second component is chosen to maximize the variance, subject to the condition of orthogonality to the first component. Each new component must be orthogonal to all previous ones, and is pointing in the direction of maximal variance, subject to these constraints.
As an example, Figure 11.1 shows how much of the variance of the SRBCT gene expression data matrix is included in the different principal components. Using the 10 first principal components will in this case include more than 60% of the variance.
In our example, we used mean centered values for the PCA and did not perform any rescaling. In principle, however, any rescaling approach is possible, since the selection of principal components as ANN inputs can be done in a supervised manner.