

Berrar D. et al. - Practical Approach to Microarray Data Analysis
.pdf154 |
Chapter 8 |
Bayesian network is used for the classification of microarray samples and for the knowledge discovery.
3.1Discretization and Selection
Discretization means to categorize the gene expression levels into several regions, e.g., ‘over-expressed’ and ‘under-expressed’. The discretization of gene expression levels before learning Bayesian networks has its own benefits and drawbacks. The multinomial model for discrete variables could represent more diverse and complex relationships than the linear Gaussian model for continuous variables because the latter could only represent the linear relationships among variables. Nevertheless, the discretization step must incur some information loss. There are various methods for discretization (Dougherty et al., 1995) and one simple method is to divide the expression level of a gene based on its mean value across the experiments. The selection step is necessary because there exist a large amount of genes that are not related to the sample classification. Generally, considering all of these genes increases the dimensionality of the problem, presents computational difficulties, and introduces unnecessary noise. So, it is often helpful to select more relevant or predictive genes for the classification task. For the selection of genes, mutual information (Cover and Thomas, 1991), P-metric (Slonim et al., 2000), or other statistical methods can be used. The mutual information between two random variables X and Y, I(X;Y), measures the amount of information that X contains about Y and is calculated as

8. Bayesian Network Classifiers for Gene Expression Analysis |
155 |
Here,  denotes the empirical probability estimated from the data. The summation is taken over all the possible X and Y values.10 In order to select the genes much related to the class variable, the mutual information between the class variable and each gene is calculated. And all genes are ranked according to the corresponding mutual information value. Then, we can select appropriate numbers of genes from this gene list. Other measures are also applied in a similar way.
denotes the empirical probability estimated from the data. The summation is taken over all the possible X and Y values.10 In order to select the genes much related to the class variable, the mutual information between the class variable and each gene is calculated. And all genes are ranked according to the corresponding mutual information value. Then, we can select appropriate numbers of genes from this gene list. Other measures are also applied in a similar way.
3.2Learning Bayesian Networks from Data
The Bayesian network learning procedure consists of two parts. The first is to learn the DAG structure of Bayesian network as outlined in Section 2. The other is to learn the parameters for each local probability distribution under the fixed structure. Parameter learning is to estimate the most likely parameter values based on the training data. As an example, consider the Bayesian network structure in Figure 8.1. We assume that each variable is binary. The value of ‘Class’ variable is either ‘class 0 (0)’ or ‘class 1 (1)’. Each gene node has the value of either ‘under-expressed (0)’ or ‘overexpressed (1)’. Then, the local probability distribution of ‘Gene G’ could be represented as depicted in Table 8.1.
The local probability distribution model of ‘Gene G’ has four parameters  These parameters could be simply estimated from the data. For example, the maximum-likelihood value of
These parameters could be simply estimated from the data. For example, the maximum-likelihood value of 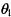 is calculated as follows:
is calculated as follows:
10 Equation 8.3 is for the case of discrete variables. If X and Y are continuous variables, then the summation in this equation can be replaced by the integral.

156 Chapter 8
When the data is not complete (some cases have missing values), the expectation-maximization (EM) algorithm 11 (Dempster et al., 1977) is generally used for the maximum-likelihood estimation of the parameter values (Heckerman, 1999).
Structure learning corresponds to searching for the plausible network structure based on the training data. The fitness of the network structure is measured by some scoring metrics. Two popular such metrics are the minimum description length (MDL) score and the Bayesian Dirichlet (BD) score (Heckerman et al., 1995; Friedman and Goldszmidt, 1999). The MDL score and the logarithm of the BD score assume the similar form and asymptotically have the same value with opposite sign. They can be decomposed into two terms i.e. the penalizing term about the complexities of the network structure and the log likelihood term. The scoring metric for the Bayesian network structure G with N variables and the training data D consisting of M cases  can be expressed as:
can be expressed as:
where  is the joint probability of
is the joint probability of  represented by the Bayesian network G. The value of
represented by the Bayesian network G. The value of is given by
is given by  and
and  is the configuration of
is the configuration of  at
at  However, the number of possible structures of Bayesian networks consisting of N variables is super-exponential in N. And it is known to be an NP-hard problem12 to find the best structure (Chickering, 1996). Hence, several search heuristics, such as greedy hill-climbing, are used to find good structures in a general way. The greedy search algorithm for learning the Bayesian network structure proceeds as follows.
However, the number of possible structures of Bayesian networks consisting of N variables is super-exponential in N. And it is known to be an NP-hard problem12 to find the best structure (Chickering, 1996). Hence, several search heuristics, such as greedy hill-climbing, are used to find good structures in a general way. The greedy search algorithm for learning the Bayesian network structure proceeds as follows.
Generate the initial Bayesian network structure  For m = 1, 2, 3, ... until convergence.
For m = 1, 2, 3, ... until convergence.
Among all the possible local changes (insertion of an edge, reversal of an edge, and deletion of an edge) in the one that leads to the largest improvement in the score is performed. The resulting graph is
the one that leads to the largest improvement in the score is performed. The resulting graph is 
11The EM algorithm proceeds as follows. First, all the parameter values are assigned randomly. Then, the following two steps are iterated until convergence. The expectation
step is to estimate the necessary sufficient statistics based on the present parameter values and given incomplete data. The maximization step is to calculate the maximum-likelihood parameter values based on the sufficient statistics estimated in the expectation step.
12The complex class of decision problems which are intrinsically harder than those that can be solved by a non-deterministic Turing machine in polynomial time.

8. Bayesian Network Classifiers for Gene Expression Analysis |
157 |
The algorithm stops when the score of  is equal to the score of
is equal to the score of  The greedy search algorithm does not guarantee to find the best solution because it will get stuck at the local maximum. Nevertheless, this algorithm has shown acceptable performance in many applications. In some situations, the greedy search algorithm with random restarts is used for escaping from the local maxima. In this algorithm, when a local maximum is found, the network structure is randomly perturbed and the greedy search procedure is applied again.
The greedy search algorithm does not guarantee to find the best solution because it will get stuck at the local maximum. Nevertheless, this algorithm has shown acceptable performance in many applications. In some situations, the greedy search algorithm with random restarts is used for escaping from the local maxima. In this algorithm, when a local maximum is found, the network structure is randomly perturbed and the greedy search procedure is applied again.
4.PROS AND CONS OF THE BAYESIAN NETWORK CLASSIFIER
4.1The Advantages of the Bayesian Network Classifier
Most advantages of the Bayesian network classifier come from its representation power. Other predictive classification models are basically focusing on learning only the relationships between the class label and input attributes. In contrast, Bayesian networks represent the joint probability distribution over the class label and input attributes. One advantage of the Bayesian network classifier is that it can predict the class label when only partial information about the input attributes is available. For example, consider again the Bayesian network in Figure 8.1. Assume that we are given a sample consisting of only the values of ‘Gene A’ and ‘Gene F’. Then, this sample could also be classified by calculating the conditional probability, P(Class | Gene A, Gene F) = P(Class, Gene A, Gene F) / P(Gene A, Gene F) from the Bayesian network. Of course, the calculation of this conditional probability is not straightforward because it requires the summation over all the possible configurations for unknown variables. If all
the variables are binary, we should enumerate |
possible |
configurations for the summation. l3 Calculation of the |
conditional |
probabilities in the Bayesian network is often called probabilistic inference. Although the probabilistic inference of arbitrary conditional probabilities from arbitrary Bayesian networks is known to be NP-hard (Cooper, 1990), there exist several algorithms applicable to the special type of network structures (Pearl, 1988; Spirtes et al., 2000; Jensen, 2001). One example is Pearl’s message passing scheme (Pearl, 1988).
Another advantage of the Bayesian network classifier is that it can be used as a hypothesis generator about the domain. The Bayesian network structure learned from microarray data represents various probabilistic relationships among gene expressions and the class label in a
13When the number of unknown variables or the range of possible values of the discrete variable is large, this problem becomes serious.
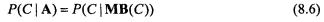
158 |
Chapter 8 |
comprehensible graph format. Such relationships might be used as a hypothesis and be verified by further biological experiments. Of course, other predictive models such as decision trees could represent the relationships between the class label and input attributes in the form of comprehensible ‘if-then’ rules. However, the representation power of the Bayesian network is superior to that of other predictive models.
4.2 Difficulties in Using Bayesian Networks for Classification
Although the Bayesian network has some advantages, it requires special tuning techniques to achieve the good classification accuracy of other predictive models in practice. In principle, this comes from the fact that Bayesian networks try to represent the joint probability distribution. When calculating the conditional probability of the class variable in the Bayesian network, only the Markov blanket (Pearl, 1988; also refer to Chapter 6 of this volume) of the class variable affects the results. For a set of N – 1 input attributes,  and the class variable C, the Markov blanket of C, MB(C), is the subset of A which satisfies the following equation.
and the class variable C, the Markov blanket of C, MB(C), is the subset of A which satisfies the following equation.
In other words, C is conditionally independent of A – MB(C) given the values of all the members of MB(C).14 Given a Bayesian network structure, determination of the Markov blanket of a node is straightforward. By the conditional independencies asserted by the network structure, the Markov blanket of a variable C consists of all the parents of C, all the children of C, and all the spouses of C. In Figure 8.1, the Markov blanket of ‘Class’ node consists of six gene nodes i.e. ‘Gene A’, ‘Gene B’, ‘Gene C’, ‘Gene D’, ‘Gene F’, and ‘Gene G’, Because only the members of the Markov blanket of the class variable participate in the classification process 15 , the construction of the accurate Markov blanket structure around the class variable is most important for the classification performance. However, the nature of the scoring metrics used in general Bayesian network learning is not favorable to this point. Consider learning the Bayesian network consisting of one class variable C and N – 1 input variables,  Then, the log likelihood term in Equation 8.5 can be decomposed into two components as
Then, the log likelihood term in Equation 8.5 can be decomposed into two components as
14Several Markov blankets could exist for a variable. The minimal Markov blanket is also called Markov boundary (Pearl, 1988). In our paper, Markov blanket always denotes the Markov boundary.
15This situation occurs when all the input attribute values are given in the sample.

8. Bayesian Network Classifiers for Gene Expression Analysis |
159 |
where  is the value of C and
is the value of C and  is the value of
is the value of  in the
in the  training example. Because only the first term of Equation 8.7 is related with the classification accuracy, maximizing the second term might mislead the search for the Bayesian network structure as a good classifier. In the greedy search procedure, the essential variable for the classification might be eliminated from the Markov blanket of the class variable. More details on this issue could be found in (Friedman et al., 1997). In the next subsection, some methods for improving classification accuracy of the Bayesian networks are briefly presented.
training example. Because only the first term of Equation 8.7 is related with the classification accuracy, maximizing the second term might mislead the search for the Bayesian network structure as a good classifier. In the greedy search procedure, the essential variable for the classification might be eliminated from the Markov blanket of the class variable. More details on this issue could be found in (Friedman et al., 1997). In the next subsection, some methods for improving classification accuracy of the Bayesian networks are briefly presented.
4.3Improving the Classification Accuracy of the Bayesian Network
There are various criteria for the classification accuracy including the total rate of correctly classified samples, the sensitivity and specificity, and the receiver operating characteristic (ROC) curve. In this chapter, we rely on the simple and intuitive measure, the total rate of correctly classified cases in the test data set.
One simple solution for the problem discussed in Section 4.2 is to fix the structure as appropriate for the classification task. The naive Bayes classifier (Mitchell, 1997) is a typical example, where all the input variables are the children of the class variable and are conditionally independent from each other given the class label. The classification performance of the naive Bayes classifier is reported to be comparable to other state-of-the-art classification techniques in many cases. However, the strong restriction on the network structure hides one advantage of the Bayesian network, that is to say, the ability of exploratory data analysis. Friedman et al. (1997) suggested the tree-augmented naive Bayes classifier (TAN) model. The TAN model assumes a little more flexible structure than the naive Bayes classifier. Here, the correlations between input variables can be represented in some restricted forms. This approach outperforms the naive Bayes classifier in some cases. Bang and Gillies (2002) deployed the hidden nodes for capturing the correlations among the input attributes. This approach has also shown the better classification accuracy although the experiments were confined to only one classification problem. Zhang et al. (2002) proposed to use the ensemble of heterogeneous Bayesian networks in order to improve the classification accuracy. This approach is based on the concept of committee machines (Haykin, 1999) and showed the improved performance
160 |
Chapter 8 |
applied to the classification of microarray data. In the next section, the experimental results of the ensemble of Bayesian network classifiers (Zhang et al., 2002) on two microarray data sets are presented.
5.EXPERIMENTS: CANCER CLASSIFICATION
5.1The Microarray Data Sets
We demonstrate the classification performance of the ensemble of Bayesian networks (Zhang et al., 2002) on two microarray data sets. These two microarray data sets are as follows.
Leukemia data: This data set is the collection presented by Golub et al. (1999).16 The data set contains 72 acute leukemia samples which consist of 25 samples of acute myeloid leukemia (AML) and 47 samples of acute lymphoblastic leukemia (ALL). 38 leukemia samples (11 AML and 27 ALL) were derived from bone marrow taken before treatment and are used as a training set in (Golub et al., 1999; Slonim et al., 2000). Additional 34 samples (14 AML and 20 ALL) were obtained as a test set among which 25 samples were derived from bone marrow and 9 were from peripheral blood. Each sample consists of 7,129 gene expression measurements obtained by a high-density oligonucleotide array. The classification task is to discriminate between AML and ALL.
Colon cancer data: This data set was presented by Alon et al. (1999) and contains 62 colon tissue samples. 40 tumor samples were collected from patients and paired 22 normal tissues were obtained from some of the patients.17 More than 6,500 human gene expressions were analyzed with an Affymetrix oligonucleotide array. Among them, 2,000 genes with highest minimal intensity across the samples were chosen (Alon et al., 1999). Finally, each sample is represented by expression levels of 2,000 genes. The classification task is to discriminate between normal tissue and cancer tissue.
5.2Experimental Settings
The P-metric (Slonim et al., 2000) was used to select 50 genes from each data set. Each gene expression level was discretized into two values, ‘overexpressed’ and ‘under-expressed’ based on its mean value across the training examples. As the scoring metric for the Bayesian network learning, BD score (Heckerman et al., 1995) with the following penalizing term was used:
16The leukemia data set is available at http://www.genome.wi.mit.edu/MPR.
17The colon cancer data set is available at http://microarray.princeton.edu/oncology/affydata.

8. Bayesian Network Classifiers for Gene Expression Analysis |
161 |
Here, N (= 51) is the number of nodes in the Bayesian network, M is the sample size,  denotes the size of a set of nodes, and
denotes the size of a set of nodes, and  denotes the number of possible configurations of a node or a set of nodes. And the ensemble machines consisting of 5, 7, 10, 15, and 20 Bayesian networks were constructed from two cancer data sets, respectively (Zhang et al., 2002).
denotes the number of possible configurations of a node or a set of nodes. And the ensemble machines consisting of 5, 7, 10, 15, and 20 Bayesian networks were constructed from two cancer data sets, respectively (Zhang et al., 2002).
5.3Experimental Results
Due to the small number of data samples in two microarray data sets used, we applied the leave-one-out cross validation (LOOCV) (Mitchell, 1997) to assess the classification performance. In the case of the leukemia data, the ensemble of seven Bayesian networks achieved the best classification accuracy (97.22%) among five ensemble machines. In the case of the colon cancer data, the ensemble machine of five Bayesian networks showed the best accuracy (85.48%) among five ensemble machines (Zhang et al., 2002).
For comparative studies, we also show the classification accuracy of other classification techniques, including weighted voting18 (Golub et al., 1999; Slonim et al., 2000), C4.5 decision trees, naive Bayes classifiers, multilayer perceptrons (MLPs), and support vector machines (SVMs) (BenDor et al., 2000; Chapter 9 of this volume). For the weighted voting scheme, the original value of gene expression level was used. The decision trees were applied to the discretized data sets. The naive Bayes classifiers, the MLPs, and the SVMs were run on both the discretized and original data sets. Table 8.2 summarizes the best classification accuracy of each method on two cancer data sets. Among all of these approaches, the SVM achieved the best classification accuracy. However, the SVM used the original gene expression values. When using discretized gene expression levels, the ensemble of Bayesian networks also shows the best performance like the SVM (in the case of leukemia data) or the MLP as well as the naive Bayes classifier (in the case of colon cancer data).
18In this approach, the classification of a new sample is based on the “weighted voting” of a set of informative genes. Each gene votes for the class depending on the distance between its expression level in the new sample and the mean expression level in each class. For more details, refer to (Golub et al., 1999; Slonim et al., 2000).

162 |
Chapter 8 |
Figure 8.3 shows the part around the class variable of a member Bayesian network which belongs to the ensemble machine learned from the leukemia data and the colon cancer data.
These graph structures generate the hypotheses on the relationships among the cancer class and gene expression profiles. These hypotheses could further guide biological experiments for the verifications. We illustrate some of them. In Figure 8.3(a), M96326, M31523, and X17042 are closely related through M84526 and ‘Cancer’ node. The E2A locus (M31523) is a frequent target of chromosomal translocations in B-cell ALL (Khalidi et al., 1999). E2A encodes two products, E12 and E47, that are part of the basic helix-loop-helix (bHLH) family of transcription factors. The disruption of E2A allele contributes to leukemogenesis (Herblot et al., 2002). Accidentally, azurocidin (M96326), adipsin (M84526), and E2A (M31523) are located in the same chromosomal region of 19pl3.3 that is known to be
8. Bayesian Network Classifiers for Gene Expression Analysis |
163 |
the site of recurrent abnormalities in ALL and AML. Among these proteins, special attention should be made to azurocidin, also known as heparinbinding protein (HBP) or CAP37, that has antibacterial properties and chemotactic activity toward monocytes (Ostergaard and Flodgaard, 1992). Azurocidin released from human neutrophils binds to endothelial cellsurface proteoglycans. A significant fraction of proteoglycan-bound azurocidin is taken up by the cells. And the internalized azurocidin markedly reduces growth factor deprivation-induced caspase-3 activation and protects endothelial cells from apoptosis (Olofsson et al., 1999). This kind of reaction might affect the behavior of leukemia in the context of cell proliferation. It might be investigated whether adipsin (M84526) plays any role in the interaction between azurocidin (M96326) and hematopoetic proteoglycan core protein (XI7042).
6.CONCLUDING REMARKS
We presented the Bayesian network method for the classification of microarray data. The Bayesian network is a probabilistic graphical model which represents the joint probability distribution over a set of variables. In the microarray data analysis, the variables denote the gene expression levels or the experimental conditions such as the characteristics of tissues and cell cycles. The Bayesian network consisting of the gene variables and the class variable can be learned from the microarray data set and used for the classification of new samples. For classification in the Bayesian network, the conditional probability of the class variable given the values of input attributes, is calculated from the joint probability representation. One of the most interesting points of the Bayesian network as a classifier compared to other predictive classification models is that it does not discriminate between the class label and the input attributes but tries to show the probabilistic dependency among arbitrary attributes. This enables the Bayesian network to represent correlations among input attributes as well as between the class variable and the input variables. Due to these features, the Bayesian network learned from microarray data could aid in broadening the knowledge about the domain by representing these relations in a comprehensible graph format. However, this flexibility of the Bayesian network makes it harder to learn the model with high classification accuracy as other classification models. This problem could be partially resolved by other techniques such as the ensemble of Bayesian networks. In our experiments on the gene expression analysis of leukemias and colon cancers, we showed that the ensemble of Bayesian classifiers could achieve the competitive classification accuracy compared to the other state-of-the-art techniques. We also demonstrated that