

Dictionary of DNA and Genome Technology
.pdfribotyping
interpretation of results on a short-term basis (a few years) [J Clin Microbiol (1999) 37:788–791].
One potential problem with this method is that samples of DNA derived directly from cells may have a pattern of modification (i.e. methylation) which is inhibitory to certain types of restriction endonuclease – thus precluding the use of these enzymes. To avoid this problem, the target sequence can be copied (from genomic DNA) by PCR and the resulting amplicons used as the sample DNA for RFLP analysis; this DNA, being synthesized in vitro, is non-methylated.
(See also PCR-RflP ANALYSIS.)
RGD motif See (β2) INTEGRIN.
rglA gene Syn. MCRA GENE. rglB Syn. MCRBC.
Rh1B helicase See DEGRADOSOME.
rho-zero (ρ0) cells Cells which lack mitochondrial DNA; they are used e.g. in the preparation of CYBRIDS.
ribavirin 1-β-D-ribofuranosyl-1,2,4-triazole-3-carboxamide: a broad-spectrum antiviral agent which is used therapeutically against e.g. the hepatitis C, Lassa fever and SARS viruses.
Ribavirin has been reported to inhibit e.g. GTP synthesis and viral RNA-dependent RNA polymerase.
One proposal was that the activity of ribavirin mimics the 7-methylguanosine CAP region on eukaryotic mRNAs. This suggestion was tested by assessing the ability of ribavirin triphosphate to interfere with the interaction between capped mRNA and eIF4E (that part of the initiation complex which recognizes the cap region); it was concluded that ribavirin did not behave as a cap analog [RNA (2005) 11(8):1238–1244].
RibEx See RIBOSWITCH.
RiboGreen® A fluorescent dye used e.g. for quantitation of RNA in solution (Molecular Probes Inc., Eugene OR/Invitrogen, Carlsbad CA). For quantitation of RNA, the sensitivity of RiboGreen is reported to be at least 1000-fold better than that obtainable by conventional ULTRAVIOLET ABSORBANCE measurement at 260 nm.
Enhancement of fluorescence is also shown when the dye binds to DNA.
(See also DNA STAINING.)
ribonuclease Any of various enzymes which cleave RNA – see under RNASE for specific examples.
Ribonucleases that have been used for sequencing RNA are mentioned in the entry MAXAM–GILBERT METHOD.
An endoribonuclease acts at internal sites while an exoribonuclease cleaves terminal nucleotides.
Ribonucleases may need to be inactivated in solutions etc. in order to prevent loss of sample RNA. Inactivation can be achieved e.g. by DIETHYLPYROCARBONATE and by products such as RNAsecure™ (Ambion, Austin TX); this commercial product can be used for treating almost any type of solution, including those containing Tris, and there is no requirement for post-treatment autoclaving.
ribonuclease protection assay A method used e.g. for detecting a specific type of mRNA in a sample that contains total RNA. Briefly, a labeled antisense oligonucleotide is allowed
to hybridize with the required mRNA in solution. Then, the addition of RNase degrades single-stranded RNA, leaving the hybridized (double-stranded) target mRNA intact. The RNase is inactivated, and the double-stranded target molecule is precipitated and is subsequently examined e.g. in a (denaturing) polyacrylamide gel.
Nucleases used in this type of assay include e.g. mung bean nuclease and endonuclease S1.
ribonuclease T1 See RNASE T1.
ribonucleotide reductase An enzyme that catalyzes the conversion of ribonucleotides to deoxyribonucleotides.
RiboPrinter Microbial Characterization System™ See entry
RIBOTYPING.
ribosomal frameshifting See FRAMESHIFTING.
ribosome density mapping (RDM) A method used for determining the number of ribosomes associated with particular regions of an mRNA molecule; the procedure involves sitespecific cleavage of polysomal mRNAs and separation of the fractions in a sucrose density gradient; the products are then analyzed [Nucleic Acids Res (2005) 33(8):2421–2432].
riboswitch A regulatory sequence within an mRNA molecule (typically in a non-coding part); on binding a given metabolite, a riboswitch undergoes a structural change that may e.g. inhibit translation. That part of a riboswitch which binds the metabolite is called an aptamer; the sequence that undergoes structural change is called the effector element or expression platform.
Riboswitches are found in prokaryotes [J Bacteriol (2005) 187(2):791–794; J Bacteriol (2005) 187(23):8127–8136], and they have also been reported in some eukaryotes (e.g. fungi, plants).
[RibEx (a web server for locating riboswitches and other conserved bacterial elements): Nucleic Acids Res (2005) 33 (Web Server issue):W690–W692.]
Online source:
RibEx: http://www.ibt.unam.mx/biocomputo/ribex.html
A system analogous to the riboswitch, but engineered, has been used to regulate gene expression in mammalian cells [RNA (2006) 12:710–716].
ribothymidine See NUCLEOSIDE.
ribotype See RIBOTYPING.
ribotyping (microbiol.) A method for TYPING bacteria which exploits inter-strain variability in the RRN OPERON (encoding 16S, 23S and 5S rRNA).
The rrn operon is useful as a target in ribotyping for three main reasons:
•it is found in all bacteria;
•most species of bacteria contain more than one copy of the rrn operon (Escherichia coli has seven copies);
•the intergenic spacers are commonly of variable length. Essentially, the genomic DNA of a given strain is cleaved
by a restriction endonuclease and the fragments are separated into bands by gel electrophoresis. A labeled DNA (or RNA) probe, complementary to a sequence in the rrn operon, is then allowed to bind to those bands of fragments containing
227

ribozyme
the target sequence. Commonly, between three and six bands are labeled by the probe. The number and position of bands produced from any given strain will depend on the following factors:
•the number of rrn operons in the genome;
•the lengths of the intergenic spacers;
•the precise distribution of cutting sites of the restriction endonuclease used in the procedure; loss or gain of recognition sites (e.g. through point mutations) will affect the fingerprint by altering the number/position of bands in the gel.
A strain defined by ribotyping is called a ribotype.
In one study on Vibrio cholerae the authors referred to high
levels of recombination between the rrn operons in cells of this species (10−5 per cell per generation); they pointed out that any given rcombinational event could undergo reversal over extended periods of time and suggested that ribotyping may not be a suitable method for determining evolutionary relationships between strains. Nevertheless, in general, ribotyping is often used successfully for differentiating strains of various species of bacteria in a short-term setting.
The study on V. cholerae referred to above also found that recombination among rrn operons often resulted in a gain or loss of cutting sites for the restriction endonuclease BglI; this has suggested a reason why the range of ribotypes detected by this enzyme is often greater than that obtained with other types of restriction enzyme.
Attempts to accelerate the ribotyping procedure have lead to the development of PCR RIBOTYPING and also to an automated approach (the RiboPrinter Microbial Characterization System™, marketed by DuPont). The RiboPrinter was used e.g. in studies on Listeria [Appl Environ Microbiol (2005) 71 (12):8115–8122] and Enterobacter [BMC Microbiol (2006) 6:15].
ribozyme Any RNA molecule which has the ability to function as a catalyst, i.e. a role similar to that usually associated with a (protein) enzyme; the roles of ribozymes include nuclease and RNA–RNA ligase.
In nature, ribozymes occur in prokaryotic and eukaryotic cells and in some viruses; in cells, they carry out functions such as the processing of RNA precursor molecules (see e.g. RNASE P) and the self-splicing of introns.
Less well known are the DEOXYRIBOZYMES.
In DNA technology, ribozymes have been used in various ways, and in various types of combination, both in vitro and
in vivo (see e.g. GENE THERAPY).
When complexed with polyethyleneimine (PEI), ribozymes form small particles which have been used for transfection studies. (See also MELITTIN.)
A ribozyme may also be linked to an APTAMER to form a structure in which the activity of the ribozyme is regulated by the binding of a specific molecule to the aptamer.
A functional HAMMERHEAD RIBOZYME has been expressed in cells of Escherichia coli from a DNA NANOCIRCLE.
Some ribozymes (e.g. one from the ciliate protozoan Tetrahymena) are thermostable, and there has been interest in the
relationship between sequence/structure and thermostability. In studies on stabilizing mutations in the Tetrahymena ribozyme, it was found that the effect on thermostability of a given mutation was highly dependent on context; thus, for example, both of the mutations A269G and A304G (close together on the ribozyme) had to be present at the same time in order to promote stability. In general, the conclusion from these studies was that mutation to stability in RNA (which may depend e.g. on the presence of rare double mutations) is less likely to occur than it is in proteins, in which stabilizing (single-site) mutations can accumulate [RNA (2006) 12(3):
387–395].
(See also ALLOSTERIC NUCLEIC ACID ENZYMES.)
rifampicin See RIFAMYCINS.
rifampin See RIFAMYCINS.
rifamycins Macrocyclic antibiotics whose target is the β subunit of (bacterial) DNA-dependent RNA polymerase – and which therefore inhibit transcription in sensitive strains. The rifamycins are active against certain Gram-positive bacteria (including mycobacteria) and some Gram-negative bacteria (e.g. strains of Brucella, Chlamydia and Legionella). In e.g.
Mycobacterium tuberculosis, resistance to rifamycins may result from mutation in the rpoB gene (which encodes the β subunit of RNA polymerase).
The rifamycins include e.g. rifampicin (= rifampin).
(See also ANTIBIOTIC and LINE PROBE ASSAY.)
right-handed helix (of DNA) A helix which, from an axial (i.e. end-on) direction, winds away from the viewer in a clockwise fashion.
rINN Recommended international non-proprietary name.
RISC See RNA INTERFERENCE. ritonavir See PROTEASE INHIBITORS.
RLGS RESTRICTION LANDMARK GENOMIC SCANNING.
RLU Relative light unit – see e.g. ACCUPROBE.
RMCE RECOMBINASE-MEDIATED CASSETTE EXCHANGE. RNA amplification (in vitro) See NUCLEIC ACID AMPLIfiCAT-
ION.
RNA chaperone See e.g. fiNOP SYSTEM. RNA degradosome Syn. DEGRADOSOME.
RNA-dependent DNA polymerase A DNA polymerase that is able to synthesize a strand of DNA on an RNA template: see
REVERSE TRANSCRIPTASE.
RNA-dependent RNA polymerase An RNA POLYMERASE that uses an RNA template; these enzymes are encoded by some types of virus and are reported to occur in certain plants [e.g. Plant Cell (1998) 10(12):2087–2101].
In at least some cases, the role of an RNA-dependent RNA polymerase may be carried out by a cellular DNA-dependent RNA polymerase in association with a viral protein [J Virol (2005) 79(13):7951–7958].
(cf. REPLICASE and TRANSCRIPTASE.)
RNA-directed DNA methylation (RdDM) A particular form
of GENE SILENCING: see e.g. SIRNA.
RNA-DNA chimeric oligonucleotide Syn. CHIMERAPLAST.
RNA editing A normal in vivo process (in animals, plants and
228

RNAlater™
some types of microorganism) in which new transcripts are generated from existing ones.
Mechanistically distinct kinds of RNA editing are carried out in different types of organism.
One example of RNA editing in mammalian cells is seen in transcripts of apolipoprotein B, a component of chylomicrons (large lipoprotein bodies produced in the intestinal mucosa). In this example, the unedited transcript encodes a protein of 4536 amino acid residues. Editing of the transcript involves an enzyme, APOBEC-1, a CYTIDINE DEAMINASE which deaminates a particular cytidine residue in the mRNA, creating a stop codon that reduces the length of the product to 2152 amino acid residues. Such deamination involves the mRNAbinding factor ACF/ASP (= APOBEC-1 complementation factor or APOBEC-1-stimulating protein); regulation of the editing of apolipoprotein B mRNA was reported to be associated with phosphorylation of ACF [Nucleic Acids Res (2006) 34(11):3299–3308].
The activity of the mammalian enzyme AID (ACTIVATION-
INDUCED CYTIDINE DEAMINASE) may be a further example
of RNA editing.
In trypanosomatids (a group of eukaryotic microorganisms) a unique type of RNA editing occurs in many or most of the mitochondrial mRNAs. This involves insertion and deletion of uridylates by a complex of proteins (see EDITOSOME) directed by small ‘guide’ RNA molecules (gRNAs). Most of the gRNAs are encoded by DNA minicircles which occur in the
KINETOPLAST.
RNA extraction (from blood) See e.g. PAXGENE BLOOD RNA
KIT.
RNA-induced silencing complex See RNA INTERFERENCE.
RNA interference (RNAi) A form of post-transcriptional gene silencing (see PTGS) found in both plant and animal cells; the phenomenon appears to have evolved as a natural antiviral response which is triggered by the presence of intracellular double-stranded RNA (dsRNA). In this process, long molecules of dsRNA (more than several hundred nucleotides) are cut into regular-sized fragments by a dsRNA-specific RNase- III-like endonuclease referred to as DICER; these fragments of dsRNA, referred to as small interfering RNA (= siRNA: see SIRNA), are reported to be about 21–28 nucleotides in length, depending e.g. on organism. One of the strands of ds siRNA, in association with a multiprotein assembly, forms the RNAinduced silencing complex (RISC); among the proteins in this complex is Argonaute 2 which, guided by the single strand of siRNA, may cleave a specific (target) mRNA molecule.
Synthetic molecules of siRNA are used for experimental gene silencing, and RNAi has been used in one approach to
GENE THERAPY. (See also SHORT HAIRPIN RNA.)
siRNAs are also involved in a phenomenon referred to as transcriptional gene silencing (TGS): see SIRNA.
Suppression of RNAi by adenovirus-associated RNA was found specifically to involve Dicer and RISC [J Virol (2005) 79(15):9556–9565].
[Therapeutic applications of RNAi: BioTechniques (2006)
40(4) supplement.]
Long dsRNA molecules also trigger the interferon response in mammalian cells. This response, involving the activity of a dsRNA-dependent protein kinase (known as PKR), causes generalized inhibition of viral and cellular protein synthesis.
RNA interference pathway The gene-regulatory pathway that involves the activity of small RNA molecules such as SIRNAs
(see RNA INTERFERENCE) and MICRORNAs.
RNA isolation Separation of total RNA, or a specified fraction of RNA, from cells; thus, for example, poly(A)-tailed mRNA molecules may be isolated from a cell lysate by adsorption to a poly(T) matrix.
Isolation of RNA from blood may be carried out e.g. with
the PAXGENE BLOOD RNA KIT.
The isolation of RNA from source material can be delayed, if necessary, for an extended period of time (e.g. weeks) by placing the material (e.g. tissue samples) in RNALATER and then storing at room temperature, 4°C or −20°C.
RNA modification pathways (a database) See MODOMICS [Nucleic Acids Res (2006) 34(Database issue):D145–D149]. Data can be accessed via:
http://genesilico.pl/modomics/
RNA PCR An early name for REVERSE TRANSCRIPTASE PCR.
RNA polymerase (RPase) Any enzyme which can polymerize ribonucleoside 5′-triphosphates on a DNA or RNA template; this term usually refers to a DNA-dependent RNA polymerase, i.e. an enzyme that uses a DNA template.
(See also RNA-DEPENDENT RNA POLYMERASE.)
In Escherichia coli, the bulk of RNA synthesis is carried out by a single multi-component RPase; in this enzyme, the three types of component (α, β, β′), together with a sigma factor protein (σ), form the functional enzyme (holoenzyme): α2ββ′σ. This enzyme is sensitive to e.g. rifampicin.
The E. coli primase (dnaG gene product) is an RPase used for synthesis of the RNA primers that are extended by DNA synthesis to form the Okazaki fragments produced during the replication of DNA. Primase is resistant to rifampicin.
RPases of members of the ARCHAEA appear to resemble the eukaryotic (nuclear) RPases (see below) rather than bacterial enzymes.
In general, eukaryotes have three multi-component nuclear RPases. RPase I (= RPase A) synthesizes most rRNA in the nucleolus. RPase II (B) synthesizes pre-mRNA. RPase III (C) synthesizes e.g. tRNAs and 5S rRNA. At least one of these enzymes also synthesizes snRNAs for pre-mRNA SPLICING. These RPases are not inhibitied by rifamycins.
In eukaryotes, the mitochondrial and chloroplast RPases are distinct from the nuclear enzymes.
RNA-recognition motif See SR PROTEINS.
RNA sequencing See e.g. MAXAM–GILBERT METHOD.
RNA splicing (pre-mRNA) See SPLICING.
RNA staining See e.g. RIBOGREEN.
(See also DNA STAINING and ULTRAVIOLET ABSORBANCE.) RNAi See RNA INTERFERENCE.
RNAlater™ An aqueous reagent (Ambion, Austin TX) that is
229

RNAqueous™ technology
used for the storage of tissue samples, and other specimens, from which RNA is to be subsequently extracted.
[Use (e.g.): PLoS ONE (2007) 2(3):e281.]
In this product, the samples may be stored at 25°C (room temperature) for up to 1 week, at 4°C for up to 1 month, and at −20°C for an indefinite period.
When the RNA is required, the sample is removed from the reagent and is treated in the same way as a freshly harvested sample.
The use of this reagent has various advantages – e.g. (i) it protects the cellular RNA from endogenous RNases, (ii) it permits extraction of RNA – at a convenient time – from samples collected at different times, and (iii) it permits overnight transport of sample(s) between laboratories (at ‘room temperature’).
RNAqueous™ technology A commercial approach (Ambion, Austin TX) used for isolating total RNA from various types of tissue or cell without the need for phenol–chloroform extraction. Essentially, tissues are disrupted by the chaotropic agent guanidinium thiocyanate – which also inactivates the endogenous nucleases. Following dilution of the lysate with an ethanol solution, the whole is transferred to a glass-fiber filter which binds RNA; three consecutive washes are used to remove proteins and DNA etc. and the RNA is then eluted.
Various products, incorporating RNAqueous™ technology, are designed for particular applications – e.g. several kits are suitable for rt-PCR, and an automated kit is suitable for highthroughput working.
(See also NUCLEIC ACID ISOLATION.)
RNase Ribonuclease – see entries below for specific examples; see also entry RIBONUCLEASE for general information.
RNase III An endoribonuclease that cleaves double-stranded RNA (e.g. in hairpin or stem–loop structures). The enzyme is involved e.g. in the processing (maturation) of rRNA from precursor molecules (see e.g. RRN OPERON) and also in the regulation of gene expression.
The (functional) RNase III of Escherichia coli is a homodimer. dsRNA is cleaved into fragments with 5′-phosphate and 3′-hydroxyl termini and short 3′ overhangs. The factors which determine cleavage sites are reported to include both structure (e.g. helix length – often two helical turns) and sequence. Based on earlier observations it was suggested that the enzyme may recognize cleavage sites by the absence of particular base pairs within discrete regions of double-helical RNA referred to as the proximal box and the distal box. More recently, specific base pair sequence elements were reported to act as positive recognition determinants. [Characterization of RNA sequence determinants and antideterminants of processing reactivity for a minimal substrate of Escherichia coli RNase III: Nucleic Acids Res (2006) 34(13):3708–3721.]
(See also DICER.)
RNase Cocktail™ A product (Ambion, Austin TX) consisting of a mixture of highly purified ribonucleases: RNase A and RNase T1; it can be used e.g. for eliminating RNA in plasmid minipreps.
RNase E See DEGRADOSOME.
(See also BL21 STAR.)
RNase H An RNase which specifically degrades a strand of RNA that is hybridized to a strand of DNA – e.g. the strand of RNA which is hybridized to fiRST STRAND cDNA. The RNase H from Escherichia coli is an endoribonuclease that forms products with 3′-hydroxyl and 5′-phosphate termini.
Exoribonuclease activity is exhibited by the reverse transcriptase of retroviruses (but is absent e.g. in a recombinant form of the enzyme from Moloney murine leukemia virus).
RNase H is used e.g. for making cDNAs. It is also used e.g. in NASBA.
RNase P An endoribonuclease, containing protein and RNA, involved in generating 5′ termini in tRNAs; its catalytic activity is mediated by the RNA. [Human RNase P: Nucleic Acids Res (2007) 35:3519–3524.]
RNase protection assay See entry RIBONULEASE PROTECTION ASSAY.
RNase T1 (RNase T1) An endoribonuclease, encoded by the fungus Aspergillus oryzae, which specifically cleaves singlestranded RNA 3′ of guanosine residues (Gp↓N), thus forming guanosine 3′-phosphate and oligonucleotides with 3′ terminal phosphate.
(See G-LESS CASSETTE and MAXAM–GILBERT METHOD.) RNAsecure™ See RIBONUCLEASE.
rne131 See BL21 STAR.
Robust-LongSAGE See SAGE.
rolling circle A mode of nucleic acid synthesis which occurs in some circular molecules of DNA and RNA.
In (ds) DNA molecules, the process begins with a NICK in one strand; the free 3′ end is then extended by a DNA polymerase, using the un-nicked strand as template. The 5′ end of the nicked strand is progressively displaced as polymerization continues.
Rolling circle synthesis occurs e.g. in many small circular bacterial plasmids, the nicking being mediated by a plasmidencoded Rep protein; plasmid-length pieces of the displaced strand may then be circularized and then converted to dsDNA plasmids.
In bacteriophage ϕX174 (which has a single-stranded, ccc DNA genome), nicking of the (double-stranded) parental replicative form (RF) is followed by rolling circle synthesis (involving the bacterial DNA polymerase); genome lengths of the resulting displaced strand are excised, circularized and converted to progeny RFs.
In phage λ, rolling circle synthesis leads to the formation of a continuous multi-genome (double-stranded) CONCATEMER that is later cut at the (repeating) cos sites into genome-sized sections; the concatemer is double-stranded because the displaced strand is used as a template on which the complementary strand is synthesized.
Rolling circle synthesis also occurs during the replication of viroids: small plant-pathogenic circular ssRNA molecules (see VIROID).
In vitro, rolling circle synthesis has been used e.g. for the
230

rTth DNA polymerase
transcription of a DNA NANOCIRCLE.
Rolling circle synthesis was also used in a rapid, sensitive method for detecting SARS virus [J Clin Microbiol (2005) 43 (5):2339–2344].
The efficiency and specificity of rolling circle amplification were reported to be improved by a mutant form of the SSB (single-strand binding protein) from the bacterium Thermus thermophilus [Nucleic Acids Res (2006) 34(9):e69].
Rom protein See COLE1. Rop protein See COLE1. Rotor-Gene™ 6000 See HRM.
RPA (1) Recombinase polymerase amplification: a rapid, isothermal method for amplifying specific sequences of DNA; it involves recombinase-mediated binding of primers to their target sequences coupled with strand-displacement synthesis of DNA [PLoS Biol (2006) 4(7):e204].
Essentially, in the presence of ATP, the primers form complexes with molecules of a recombinase (the UvsX enzyme of phage T4), and primer–recombinase complexes search the double-stranded sample DNA for sequences complementary to the primers. When primer-binding sites are located the recombinase mediates strand exchange: the binding of a given primer displaces a loop of ssDNA which is stabilized by the binding of single-strand binding proteins (SSBPs). The disassembly of recombinase permits the bound primer to be extended by a (strand-displacing) DNA polymerase. As primer extension continues, SSBPs stabilize the increasingly long strand of displaced DNA. Similar, repeated activity by both (opposing) primers leads to exponential amplification of the target. Ongoing activity in RPA depends on the presence of agents which mediate a balance in the development and disassembly of the primer–recombinase complexes.
Reactions in which the template is either absent or in low concentration were found to lead to primer-dependent artefacts. To combat this, the detection of products is achieved by a probe-based system. Essentially, the probe includes an abasic-site mimic flanked by a fluorophore-labeled nucleotide and a quencher-labeled nucleotide; the intact probe is associated with low-level fluorescence. Hybridization of the probe to its complementary sequence permits the abasic-site mimic to be recognized – and cleaved – by an endonuclease (Escherichia coli endonuclease IV; Nfo) which subsequently results in a measureable rise in fluorescence (as the fluorophore is no longer quenched). The increase in fluorescence is exploited in monitoring the products. In that cleavage by Nfo requires stable pairing between the probe and its binding site, this arrangement promotes specificity in product detection.
RPA is reported to be capable of multiplex operation, the method having been employed e.g. to amplify simultaneously targets from several different strains of methicillin-resistant
Staphylococcus aureus (MRSA).
(2) Replication protein A (see e.g. OKAZAKI FRAGMENT).
RPase RNA POLYMERASE. rpmI gene See OPERON. rpoB gene See RIFAMYCINS.
rpoH gene See ESCHERICHIA COLI (table).
rpsL gene (syn. strA gene) In Escherichia coli: a gene encoding the ribosomal protein S12; mutation in this gene may lead to resistance to the antibiotic streptomycin (for which S12 is the target protein).
RRE Rev protein response element: see REV PROTEIN.
rrn operon (in bacteria) An OPERON encoding the three species of rRNA (16S, 23S, 5S) which is transcribed (as a single prerRNA transcript) in the order 16S→23S→5S. The genes are not contiguous: a so-called intergenic spacer region (ISR) is found between each pair of coding sequences. The length of an ISR can vary, even in different copies of the rrn operon in the same chromosome, and this feature has been exploited in
certain forms of TYPING (PCR-RIBOTYPING and RIBOTYPING).
Bacteria usually contain multiple copies of the rrn operon, but single copies are reported to occur e.g. in Mycobacterium tuberculosis and Tropheryma whipplei.
Escherichia coli contains seven copies of the rrn operon. The pre-rRNA molecule forms secondary structures (through base-pairing between inverted repeats) in which the 16S and 23S components form loops in separate stem–loop structures; the 16S and 23S precursor molecules are liberated by the action of an endoribonuclease (RNase III).
(See also LEUCINE-RESPONSIVE REGULATOR PROTEIN.) RT-PCR REVERSE TRANSCRIPTASE PCR.
rt-PCR REVERSE TRANSCRIPTASE PCR.
Rta (brlf-1 gene product) See ZEBRA.
rtPCR REVERSE TRANSCRIPTASE PCR.
rTth DNA polymerase A 94-kDA recombinant DNA polymerase (Perkin Elmer/Applied Biosystems) that is derived from the thermophilic bacterium Thermus thermophilus; in a single buffer system it can act as both a reverse transcriptase and a DNA-dependent DNA polymerase.
The enzyme can function in the range 60–70°C and is used
e.g. in REVERSE TRANSCRIPTASE PCR.
Examples of use: studies on the SARS virus [BMC Infect Dis (2006) 6:20] and studies on the hepatitis C virus [J Clin Microbiol (2006) 44(7):2507–2511].
231

S
S (1) A specific indicator of ambiguity in the recognition site of a RESTRICTION ENDONUCLEASE or in any other sequence of nucleic acid; thus, for example, in ↓GTSAC (enzyme Tsp45I) the ‘S’ indicates G or C.
(2) L-Serine (alternative to Ser).
S1 nuclease Syn. ENDONUCLEASE S1.
Saccharomyces A genus of YEASTS; the species S. cerevisiae is widely used as an experimental organism (and is also used in the manufacture of alcoholic beverages and fermented products).
S. cerevisiae occurs as single cells which may be haploid or diploid, depending on the stage of the life cycle. Both haploid and diploid cells reproduce asexually by a process known as
budding (cf. SCHIZOSACCHAROMYCES).
Sexual reproduction involves fusion of two haploid cells of appropriate MATING TYPE. The diploid zygote gives rise to diploid somatic (vegetative) cells. Diploid cells constitute a major phase in the life cycle. Diploid cells undergo meiosis to form four ascospores within a structure called an ascus; two of the ascospores are of the α mating type, and two are of the a mating type. Ascospores develop into haploid somatic cells.
The existence of both haploid and diploid somatic cells in the life cycle has been exploited in complementation studies in which e.g. the diploid cells can be used to study the effects of different alleles derived from parental haploids.
Cells of the AB1380 strain of S. cerevisiae, and derivatives, have been used e.g. as hosts for YEAST ARTIfiCIAL CHROMO- SOMES. These cells have mutations in genes TRP1 and URA3 and require both tryptophan and uracil in growth media; the corresponding (wild-type) genes in YACs are therefore useful for the selection of YAC-containing cells (in tryptophandeficient and uracil-deficient media). The cells of this strain also have a mutation in a gene whose product is involved in adenine metabolism, leading to red pigmentation; this defect is bypassed by the suppressor gene SUP4 which, in a YAC, is involved in a mechanism for indicating the presence/absence
of an insert (see YEAST ARTIfiCIAL CHROMOSOME).
S. cerevisiae is used in the YEAST TWO-HYBRID SYSTEM for
studying protein–protein interactions.
(See also ARS and YEAST MARKER.)
[Yeast-based technologies in genomics and proteomics (a review): BioTechniques (2006) 40(5):625–644.]
Saccharomyces cerevisiae Genome Deletion Project See entry
GENOME DELETION PROJECT.
SafeBis DNA DNA which has been treated with BISULfiTE but which has not been subsequently desulfonated; it has been used e.g. in studies on DNA methylation [Nucleic Acids Res (2007) 35(1):e4].
safety cabinet (syn. sterile cabinet) A piece of equipment that provides a partially or totally enclosed space within which certain types of procedure can be carried out; some types of cabinet are designed specifically to provide effective contain-
ment of dangerous pathogens – with the object of protecting both laboratory staff and the environment.
Class I cabinets are accessed, for working, via an opening in the front below a glass viewing panel. A fan draws air into the cabinet, via the front opening, and air leaves the cabinet via a high-efficiency particulate air filter (a HEPA filter); the rate of airflow is such that aerosols do not leave the cabinet via the front opening.
Class I cabinets are used e.g. for work on pathogens such as
Coxiella burnetii, Francisella tularensis and Mycobacterium tuberculosis. In addition to the use of a class I cabinet, work with this type of pathogen requires the use of gloves and also the existence of plenum ventilation in the laboratory – i.e. a constant inflow of air to the laboratory, with filtration of the exhausted air through a HEPA filter.
Class II cabinets are used primarily to protect materials in the cabinet from environmental contamination. Filtered air constantly flows down onto the work surface and leaves the cabinet after further filtration. Access is from the front, below a glass viewing panel. These cabinets (which are common in microbiological laboratories in universities) are used e.g. to avoid contamination when inoculating media.
Class III cabinets are high-security cabinets that are totally enclosed and gas-tight. Air is filtered before entry and before discharge. Manipulation of materials etc. within the cabinet is conducted by means of arm-length rubber gloves that are fitted securely into the front of the cabinet. The interior of the cabinet is viewed via a glass panel. Access to the cabinet is via a separate two-door sterilization/disinfection chamber.
Class III cabinets are used when handling hazardous pathogens such as the Ebola virus, Lassa fever virus and Marburg virus.
SAGE Serial analysis of gene expression: a method for detecting, identifying and quantitating genes expressed by a given type of cell, under given conditions, by analyzing a collection of short (~10-nucleotide) sequences (SAGE tags, diagnostic tags) derived from the transcript (mRNA) of each gene. This method may be used without prior knowledge of the nucleotide sequences of genes; a particular gene may be identified via its diagnostic tag.
SAGE is used for discovering new genes, for studies on the upand down-regulation of genes under different conditions, for studies on differential expression of genes, and for establishing links between various aspects of a cell’s physiology. Thus, for example, observation of the levels of transcripts has revealed a relationship between phosphate metabolism and other aspects of cell physiology in the smut fungus Ustilago maydis [Eukaryotic Cell (2005) 4(12):2029–2043], and the use of LongSAGE (see later) has facilitated gene discovery in a marine coccolithophorid [Appl Environ Microbiol (2006) 72:252–260].
The basic procedure for SAGE is essentially as follows. Initially, total mRNA is isolated from the cell or tissue and
233

salicylate
is reverse transcribed, with BIOTINylated oligo(dT) primers, to cDNAs.
Double-stranded cDNA molecules are then subjected to a ‘frequent-cutting’ restriction endonuclease (e.g. NlaIII) which cleaves the cDNA molecules more than once. (This enzyme is sometimes called the ‘anchoring enzyme’.)
The biotinylated ends of the cleaved cDNA molecules are then captured e.g. by STREPTAVIDIN-coated DYNABEADS.
The Dynabead-bound fragments are then separated into two subpopulations.
In one of the subpopulations, the free ends of the fragments (i.e. termini formed by cleavage with the anchoring enzyme) are ligated to a dsDNA oligonucleotide linker (linker A); this linker contains the recognition site of a type IIS RESTRICTION
ENDONUCLEASE.
In the second subpopulation, the free ends are ligated to a different linker (linker B); however, this linker also contains the recognition site of the type IIS restriction enzyme.
Each subpopulation is subjected to the action of a type IIS restriction enzyme. This type of enzyme cuts some distance from its recognition site; consequently, the result of type IIS restriction is the generation of a number of free fragments (released from the Dynabeads), each of which consists of (i) the linker, and (ii) a short sequence of nucleotides (9–14 bp) derived from the (contiguous) ligated cDNA fragment. The sequence of nucleotides derived from the cDNA fragment is called the signature tag for the relevant gene.
Any overhang produced by type IIS restriction is filled in e.g. by the Klenow fragment.
The two subpopulations are mixed.
The tags are now ligated together by blunt-ended ligation. Structures of the following type are formed:
linker A–(signature tag)2–linker B
The pair of ligated signature tags is called a ditag. The ditags are amplified by PCR, using primer-binding sites in each of the two types of linker.
The PCR products are subjected to the restriction enzyme used initially to cleave the cDNAs (the anchoring enzyme). This results in the excision of both (terminal) linkers, leaving a pair of ligated signature tags (a ditag).
The ditags are ligated together, serially, to form long molecules of DNA, each of these molecules containing many tags. These molecules can be cloned and sequenced.
The relative abundance of a given transcript in the original sample is reflected in the number of relevant tags in the final product. A signature tag identifies a particular gene – so that the level of expression of a given gene can be assessed by the SAGE procedure.
Computer-based analysis of the sequence data can be used to identify particular genes.
Various modifications of the basic scheme described have been devised:
MicroSAGE uses minute quantities of sample material. The
principle is similar to that described, although e.g. mRNAs in the sample bind directly to oligo(dT)-coated magnetic beads (i.e. prior to reverse transcription).
LongSAGE [Nat Biotechnol (2002) 20(5):508–512] uses a longer signature tag (21 bp), and Robust-LongSAGE offers a number of improvements [Plant Physiol (2004) 134(3):890– 897].
(See also 5′ LongSAGE and 3′ LongSAGE [Proc Natl Acad Sci USA (2004) 101(32):11701–11706].)
In superSAGE [Proc Natl Acad Sci USA (2003) 100(26): 15718–15723], a type III restriction endonuclease (EcoP15I) is used to increase the size of tags to 26 bp; superSAGE e.g. facilitates the identification of a gene from databases such as GenBank.
salicylate (as an inducer of gene expression) See e.g. CASCADE
EXPRESSION SYSTEM.
Salmonella A genus of Gram-negative, rod-shaped, generally motile bacteria of the family Enterobacteriaceae (see entry ENTEROBACTERIA); the species S. typhi is the causal agent of typhoid.
GC% of chromosomal DNA: 50–52.
The salmonellae (i.e. members of this genus) can be grown on (e.g.) nutrient agar and MacConkey’s agar; media used for enrichment include e.g. selenite broth and tetrathionate broth.
Linguistic note
The name Salmonella derives from the name of an American bacteriologist, D. E. Salmon; the correct pronunciation of the genus name is therefore ‘Salmon-ella’ – the first ‘l’ is silent.
Salmonella/microsome assay Syn. AMES TEST.
SAM S-adenosyl-L-methionine (a methyl donor). same-sense mutation Any mutation which fails to change the
identity of the amino acid that is specified by a given codon. A same-sense mutation may not be equivalent to a SILENT
MUTATION if it introduces a CODON BIAS.
SAMPL Selective amplification of microsatellite polymorphic loci, or selectively amplified microsatellite polymorphic loci: a method for detecting variations (in length) of microsatellite sequences in genomic DNA. Initially – as in AflP – genomic DNA is digested with two restriction enzymes, the fragments are ligated to adaptors, and pre-amplification is carried out by PCR with primers having a single selective 3′ nucleotide.
Another round of PCR amplification is carried out with one AFLP primer and a (labeled) SAMPL primer complementary to the particular microsatellite sequence being studied; the two primers in the second round of PCR therefore amplify those microsatellite sequences that were present in amplicons formed during pre-amplification.
sampling (DNA) See, for example, BUCCAL CELL SAMPLING
and FORENSIC APPLICATIONS.
Sandhoff disease A GENETIC DISEASE similar to TAY–SACHS
DISEASE (q.v.) but resulting from a deficiency in the activity of the β-subunit of hexosaminidase A.
Sanger’s method (DNA sequencing) See DIDEOXY METHOD.
Sapphire-II™ See CHEMILUMINESCENCE ENHANCER. saquinavir See PROTEASE INHIBITORS.
234

SDA
SARS virus (detection) A rapid, sensitive method for detecting the SARS virus (an ssRNA coronavirus) in clinical samples has exploited (genome-specific) PADLOCK PROBES which, on circularization, act as templates for isothermal replication of target sequences by the ROLLING CIRCLE MECHANISM [J Clin Microbiol (2005) 43(5):2339–2344].
SBE See SINGLE-BASE EXTENSION.
scaffold In a (eukaryotic) chromosome: a central, proteinaceous structure which serves to anchor loops of DNA.
scaffolding proteins Proteins that form a temporary (structural) framework during the assembly of (e.g.) a phage head – but which do not form part of the mature (completed) head. The scaffolding proteins may be subsequently degraded – as e.g. in phages λ and T4; they are re-cycled e.g. in phages P22 and ϕ29.
scanning force microscopy Syn. ATOMIC FORCE MICROSCOPY.
SCCmec In strains of MRSA: a mobile genetic element (name: staphylococcal cassette chromosome mec) which includes the MECA GENE complex and the ccr gene complex.
Different versions of the mec and ccr gene complexes have been found in environmental isolates of MRSA, and SCCmec elements have been classified into five types (I–V) on the basis of these differences; however, this classification is not ideal, and a new system of classification, which is believed to be more suitable for typing these strains, has been proposed [Antimicrob Agents Chemother (2006) 50(3):1001–1012].
SCE Single-cell ELECTROPORATION (q.v.). SCF (stem cell factor) See KIT.
scFV A SINGLE-CHAIN VARIABLE FRAGMENT.
Schizosaccharomyces A genus of fungi in which the morphological forms are single cells that reproduce by fission (rather than budding: cf. SACCHAROMYCES) or hyphae that fragment into spores. Some strains of these fungi are homothallic (i.e. self-fertile); others are heterothallic.
Studies on the effects of mutations in replication-initiation genes and checkpoint genes in the species S. pombe found that the resulting replication stress, and inappropriate mitosis, resulted in the production of reactive oxygen species (ROS) and cell death [J Cell Sci (2006) 119(1):124–131].
The species of Schizosaccharomyces are distinguished e.g. by the number of ascospores formed in the asci and by the ability to ferment specific sugars.
schlieren system A system used for representing a refractive index gradient.
Schneider’s medium A liquid medium used for the culture of insect cells; it includes various inorganic salts (e.g. magnesium sulfate, potassium chloride, sodium chloride), D-glucose, trehalose, malic acid, succinic acid, and a number of amino acids.
SCID See GENETIC DISEASE (table). SCIDX1 See GENETIC DISEASE (table).
scintillation proximity assay See SPA.
SCNT Somatic cell nuclear transfer: removal of the chromosomes from an unfertilized oocyte (enucleation) and their replacement with chromosomes from a somatic cell.
An oocyte which has undergone SCNT may be used e.g. to produce an embryonic clone which may serve as a source of embryonic stem cells (see STEM CELL). As an alternative, an SCNT-treated oocyte may be allowed to develop into a live animal (a clone) following appropriate follow-up procedures.
Scorpion probe A PROBE analogous to a MOLECULAR BEACON
PROBE used for monitoring the products of PCR [e.g. Nucleic Acids Res (2000) 28(19):3752–3761]. The probe resembles a molecular beacon, with fluorophore and quencher in the stem section – but it has a single-stranded 3′ extension. During the (heat) denaturation stage, both the target DNA and the stem section of the probe become single-stranded; on cooling, the probe’s 3′ extension binds to target DNA and is subsequently extended, copying the target sequence. During the subsequent denaturation and cooling the loop region of the probe hybridizes with a complementary sequence in the probe’s extended 3′ region; in this arrangement, the fluorophore and quencher are separated – so that the probe exhibits fluorescence under suitable excitation.
ScRad51 The RAD51 protein of Saccharomyces cerevisiae. screening Any selective procedure in which individual cells or
molecules with given characteristics are distinguished and/or isolated from the other individuals in the same population.
For examples of screening see BLACK–WHITE SCREENING, blue–white screening (in PBLUESCRIPT), COLONY HYBRID-
IZATION, ELISPOT, METAGENOME, RECACTIVE, SIGEX, SSCP ANALYSIS.
screening test (immunol., microbiol.) (1) Any test that is used to examine specimen(s) in order to detect a particular type of antibody, antigen or microorganism. Screening tests are often simple and inexpensive but are frequently not highly specific; as a positive result in a screening test may be followed-up by other – more specific – test(s), a proportion of false-positive results may be tolerated.
Examples of nucleic-acid-based screening tests include e.g.
AMP CT and LINE PROBE ASSAY.
(2) Any test designed to assign a given, unknown organism to one of a possible range of categories or taxa.
SCS system Separate-component-stabilization system: see CCD
MECHANISM.
SDA Strand displacement amplification: an isothermal method for copying a given sequence of nucleotides in a sample of DNA. (See also NUCLEIC ACID AMPLIfiCATION.) Originally, SDA was carried out at an operating temperature of ~40°C; later, the operating temperature was raised to 50°C or above (thermophilic SDA, tSDA) with the object of improving the specificity of the method.
SDA (which is shown diagrammatically in the figure) is characterized by two distinctive features: (i) the use of a particular type of RESTRICTION ENDONUCLEASE that repeatedly nicks a chemically modified recognition sequence in doublestranded intermediate forms, thereby continually regenerating sense and antisense strands that are displaced from dsDNA intermediates by a strand-displacing DNA polymerase; (ii) use of a strand-displacing DNA polymerase (such as Klenow
235
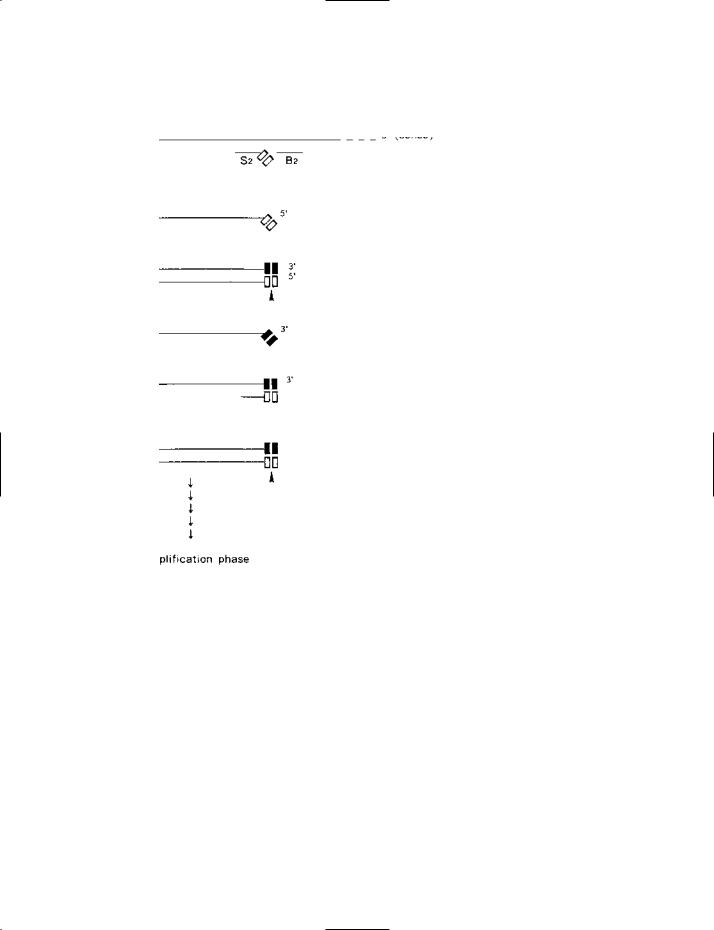
Part I
SDA (strand displacement amplification) – part I: the target-generation phase (diagrammatic).
Target DNA is initially denatured to the single-stranded state prior to SDA.
(a)The antisense (3′-to-5′) strand of the target duplex is shown with the amplicon (i.e. the sequence of nucleotides to be amplified) delimited by two short vertical bars. For clarity of presentation, and economy of space, only one strand of the target DNA is
236

SDS–PAGE
fragment: see DNA POLYMERASE I) which is able to displace a strand from duplex DNA by extending an existing 3′ terminus at the site of a nick.
Restriction endonuclease. The restriction endonuclease used in SDA is HincII, and the specific recognition site employed is:
5′-GTT/GAC-3′
3′-CAA/CTG-5′
Normally, HincII makes a double-stranded, blunt-ended cut, as shown by the oblique (/ ), but this activity does not occur in SDA because, in each recognition site, one of the two strands (n n in the figure) is chemically modified and cannot be cut; the other strand (n n in the figure) is nicked by HincII.
The nickable strand within a recognition site can be formed either by a primer or by regeneration of the recognition site by 3′ extension from a nick. Both primers, S1 and S2, have a 5′ terminal tag: 5′-GTTGAC-3′; this unmodified sequence, when present in a (duplex) recognition site, can be nicked by HincII between the ‘T’ and ‘G’ nucleotides.
The un-nickable strand in a recognition site arises because, in SDA, all strand synthesis occurs in a reaction mixture that contains a chemically modified form of deoxyadenosine triphosphate: α-thiophosphoryl dATP (dATPαS). As a consequence of the presence of dATPαS, when the complementary strand is synthesized on a template strand containing a primer
sequence, the newly synthesized strand (3′-CAACTG..........) includes two modified nucleotides in the AA positions which are adjacent to the cleavage site; because of the presence of these two modified nucleotides, this strand cannot be cleaved by HincII. Thus the double-stranded recognition site contains a nickable site (in the primer sequence) and a non-cleavable sequence in the newly synthesized (complementary) strand. This ‘hemi-modified’ HincII site (= hemiphosphorothioate site) is therefore susceptible to nicking, only, by HincII in the primer strand between ‘T’ and ‘G’ (see e.g. (e) in part I of the figure).
A nick between the ‘T’ and ‘G’ in the primer sequence of a recognition site is followed by extension from the 3′ end of the nick – this displacing the strand downstream of the nick. Importantly, the strand which is formed by such extension (5′-GTT→→) incorporates a modified nucleotide (dATPαS) at the ‘A’ site within the sequence GAC. Despite this, the HincII site that is regenerated in the newly synthesized strand (i.e. 5′-GTTGAC....3′) is nickable because the one modified nucleotide in GAC does not interfere with nickability of the sequence GTTGAC.
SDS SODIUM DODECYL SULFATE.
SDS–PAGE (Laemmli electrophoresis) Sodium dodecyl sul- fate–POLYACRYLAMIDE gel electrophoresis: a particular form of GEL ELECTROPHORESIS that is used e.g. for determining the molecular weight of a protein or separating a mixture of proteins.
SDA (continued )
considered here; corresponding events occur on the complementary strand. A primer (S1) has bound at the 3′ end of the amplicon. The 5′ end of this primer carries a tag that includes one strand of the HincII recognition sequence (5′-GTTGAC-3′) (shown by the symbol ). A bumper primer (B1 – see entry) has bound upstream of primer S1. Extension of S1 will produce a sense strand on the antisense template; this sense strand will be displaced by the extension of B1.
(b)The sense strand which has been displaced, by bumper primer B1, from the original template strand.
(c)Primer S2 has bound to the 3′ end of the amplicon on the sense strand. Like primer S1, its 5′ end is tagged with the recognition sequence of HincII. The bumper primer B2 has bound upstream of primer S2. Extension of S2 will produce an antisense copy of the amplicon which is tagged at both ends with a (single-stranded) HincII recognition sequence; this strand will be displaced by bumper primer B2.
(d)The antisense strand which has been displaced from stage (c) by bumper primer B2. Notice that the HincII sequence at the 3′ end of this strand, having been synthesized with dATPaS (see entry), is ‘modified’ (as indicated by the symbol nn); this (modified) HincII
sequence (when subsequently part of a double-stranded amplicon) is not susceptible to the restriction endonuclease HincII. At the other (5′) end of the strand, the HincII sequence is not modified because it originated in primer S2 (which was synthesized with normal nucleotides). Primer S1 (not shown) can bind at the 3′ end of this strand – the HincII sequence in S1 hybridizing with the mod-
ified sequence in the strand. Extension of S1 yields the double-stranded amplicon shown at (e).
(e)Each end of this amplicon has a ‘hemi-modified’ (hemiphosphorothioate) recognition sequence for HincII. A hemi-modified site can be nicked in the non-modified strand (as shown by the arrowheads). Nicking can therefore occur in the ‘upper’ strand or the ‘lower’
strand of this (double-stranded) amplicon. For clarity, nicking in only the upper strand is considered here. Nicking is followed by extension of the 3′ end of the nick – resulting in displacement of the nicked strand, which is shown at (f).
(f)The strand displaced from (d) following nicking and extension of the 3′ end of the nick.
(g)Primer S2 has bound to the displaced strand and will be extended to form the double-stranded product shown at (h).
(h)This product feeds into the amplification phase (see part II of the figure).
In the above scheme, at stage (e), nicking of the upper strand, only, was considered. Nicking of the lower strand, and its displacement, produces a single-stranded product that binds primer S1; extension of primer S1 leads to the formation of a double-stranded product that feeds into the amplification phase.
237