

эндокринка
.pdf4. Подавляет окисление аминокислот.
IV. Участие инсулина в регуляции водно-электролитного обмена:
1.Усиливает поглощение мышцами и печенью калия.
2.Снижает экскрецию натрия мочой.
3.Способствует задержке воды в организме.
Не вдеваясь в подробности, отметим, что в настоящее время известны, по крайней мере, 4 механизма реализации внутриклеточных эффектов инсулина:
1.Активация или торможение клеточных ферментов.
2.Активация транспорта глюкозы и аминокислот в клетки.
3.Влияние на синтез РНК и белков.
4.Ингибирование клеточной аденилатциклазы и понижение содержания внутриклеточного цАМФ.
Сам по себе механизм действия инсулина выглядит следующим образом. Инсулин связывается с рецепторами на поверхности клеток-мишеней, в первую очередь, инсулин зависимых тканей – печени, мышц, жира, соединительной ткани. На цитоплазматических мембранах вышеперечисленных тканей, как оказалось, таких рецепторов очень много – на каждой клетке от 5000 до 250000, хотя реально функционирует лишь около 10%. После подобного взаимодействия изменяются характеристики цитоплазматических мембран, и облегчается транспорт глюкозы и аминокислот. Одновременно происходит ингибирование аденилатциклазы плазматической мембраны, снижается содержание цАМФ, в результате чего запускается каскад анаболических реакций клетки: гликогенез,
синтез белков, жиров и нуклеиновых кислот.
Секреция инсулина стимулируется следующими веществами:
1) глюкоза, 2) аминокислоты, особенно аргинин и лизин, 3) бомбезин, 4) гастрин, 5) панкреазимин, 6) секретин, 7) глюкокортикоиды, 8) глюкагон, 9) адренокортикотропный
гормон, 10) соматотропный гормон, 11) -адреностимуляторы.
Тормозят выработку инсулина (1) гипогликемия, (2) соматостатин, (3) никотиновая кислота, (4) -адреностимуляторы
Второй гормон поджелудочной железы – глюкагон представляет собой одноцепочечный полипептид, состоящий из 29 аминокислотных остатков с молекулярной массой около 3500 D. В чистом виде глюкагон был выделен в 1951 году Геде. Его содержание в крови здоровых людей натощак близко к 75-150 нг/л (активны лишь 40% гормона). На протяжении суток он непрерывно синтезируется -клетками островков Лангерганса. Секреция глюкагона тормозится глюкозой и соматостатином. Как указывалось, глюкагон стимулирует липолиз, кетогенез, гликогенолиз, глюконеогенез, что ведет к повышению содержания глюкозы в крови. Существенное значение в регуляции гликемии имеет его стимулирующее действие на секрецию инсулина – косвенная стимуляция через гипергликемию и быстрая прямая стимуляция. Гормон разрушается в почках.
Механизм действия глюкагона сводится к активации через специфические рецепторы цитоплазматических мембран аденилатциклазы главным образом печени и последующего повышения содержания цАМФ в клетках. Это и приводит к гипергликемии, липолизу, кетогенезу и некоторым другим эффектам.
Основными проявлениями сахарного диабета являются следующие:
1)гипергликемия (уровень глюкозы в крови выше 6,66 ммоль/л),
2)глюкозурия (содержание глюкозы в моче может достигать 555-666 ммоль/л, за сутки в первичную мочу здоровых людей фильтруется до 150 г глюкозы, больных сахарным диабетом - около 300-600 г, а возможная потеря глюкозы мочой достигает 300 г/сутки),
3)полиурия (суточный диурез выше 2 л, но может достигать и 12 л),
4)полидипсия – жажда (прием жидкости более 2 л в сутки),
5) гиперлактацидемия (содержание лактата в крови более 0,8 ммоль/л, чаще 1,1-
1,4 ммоль/л),
6) гиперкетонемия – повышенное содержание в крови кетоновых тел (чаще выше
520мкмоль/л), кетонурия,
7)липемия (повышенное содержание в крови липидов, чаще выше 8 г/л),
8)быстрое похудание, свойственное больным с инсулин-зависимым сахарным
диабетом.
9)понижение толерантности организма к глюкозе, определяемой с помощью нагрузочной пробы глюкозой (75 г глюкозы и стакан воды, далее наличие двукратного превышения содержания глюкозы около 11,1 ммоль/л на протяжении 60-ой, 90-ой и 120ой минутах определения).
Этиология сахарного диабета. Причиной сахарного диабета является инсулиновая недостаточность. Инсулиновая недостаточность может быть панкреатической, т.е. обусловлена нарушением биосинтеза и выделения инсулина из В- клеток островков Лангерганса поджелудочной железы – так называемый инсулинзависимый сахарный диабет (ИЗСД), и внепанкреатической, которая может иметь место при нормальном выделении инсулина В-клетками – инсулиннезависимый сахарный диабет (ИНСД). Наиболее вероятной причиной ИЗСД считается генетическая предрасположенность, которая определяется, как минимум, двумя генами в шестой хромосоме. Однако, как стало сейчас известно, около 20 хромосомных участков, проявляющих признаки положительной сцепленности с заболеванием, имеют значение в развитии сахарного диабета. Их связывают с генами 2, 6, 10, 11, 14, 15, и 18 хромосом.
Генетическая предрасположенность к ИЗСД ассоциируется с определенными
антигенами лейкоцитов (система HLA). Так, распространенность антигена B8 и B15 среди больных ИЗСД составляет 49 и 21%, среди здоровых – 31 и 10%, соответственно. Поэтому
наличие у людей гаплоидов B8 или B15 увеличивает риск заболеть ИЗСД в 2-3 раза, а их сочетание, т.е. В8 + B15 – на один порядок (так называемые маркеры сахарного диабета). Однако среди системы HLA есть антигены, которые оказывают противоположное
действие, препятствуя возникновению ИЗСД. Так, гаплотип В7 обнаружен среди 13% больных, а среди здоровых – в 27% случаев. Относительный риск заболеть ИЗСД у
носителей В7 в 14,5 раза ниже, чем лиц, у которых он отсутствует. Лейкоцитарные антигены гистосовместимости определяют иммунологический характер ответа организма на различные антигены и непосредственного отношения к углеводному обмену не имеют. Таким образом, HLA-фенотип при ИЗСД является генетической детерминантой, определяющей чувствительность В-клеток поджелудочной железы к вирусным и другим антигенам и отражает характер иммунологического ответа организма.
Помимо наследственной предрасположенности этиологическими факторами сахарного диабета являются поражения самой поджелудочной железы. Известно, что большая часть из общего количества островков Лангерганса (а их чуть больше 1 млн.) приходится на хвостовую часть и тело железы. Поэтому травмы, нарушения кровообращения, инфекции и интоксикации именно этой части поджелудочной железы приводят к развитию сахарного диабета.
Согласно современным представлениям, наиважнейшим диабетогенным фактором риска является аутоиммунный процесс. В результате иммунологических реакций
образуются антитела (иммуноглобулины) и сенсибилизированные Т-лимфоциты против - клеток островкового аппарата и инсулина, что подтверждается наличием в островках особых морфологически выраженных проявлений очагов воспаления, получивших наименование инсулитов. Повреждающим эффектом обладают моноциты, макрофаги, Т- цитотоксические лимфоциты, NK-клетки, антитела (главным образом Ig G-класса), а также цитокины – инерлейкин-1, фактор некроза опухоли- , -интерферон. В 80-100% случаев у больных сахарным диабетом находят проявления гиперчувствительности немедленного и/или замедленного типов.
В качестве этиологического фактора ИЗСД могут выступать вирусные инфекции,
поражающие В-клетки островков. Особое значение придается вирусам кори, краснухи, эпидемического паротита, ветряной оспы, не Коксаки, инфекционного мононуклеоза, гепатита, цитомегаловируса, реовирусы, энтеровирусы, вирус Эпштейна-Барр. Инсулиты обнаруживаются не только при аутоаллергических заболеваниях, но и при вирусных инфекциях, которые инициируют образование в организме больных сахарным диабетом неоантигенов (вторичных аутоантигенов). Высвобождающиеся в процессе аллергических реакций из макрофагов и лимфоцитах интерлейкин-1, фактор некроза опухоли- , - интерферон и другие медиаторы аллергии обладают по отношению к -клеткам цитотоксическим, антипролиферативным и антисекреторным действиями. В результате длительного деструктивного процесса к моменту развернутой клинической картины сахарного диабета 85-95% всех -клеток уже разрушены, что приводит к абсолютной инсулиновой недостаточности.
Альтернативным фактором риска сахарного диабета может быть действие токсических для В-клеток веществ, так называемых химических диабетогенов. Так, экспериментальный сахарный диабет можно вызвать введением в организм аллоксана (аллоксановый диабет), дитизона, стрептозотоцина, нитрозаминов, нитрозомочевины. Химическими диабетогенами являются мочевая кислота, лекарственный препарат пентамидин, средство для борьбы с грызунами вакор, продукты, содержащие пищевые цианиды (африканский корнеплод кассава, которым питается около 400 млн. аборигенов). В последнее время внимание исследователей привлекают химические диабетогенные потенции белка коровьего молока – бычьего сывороточного альбумина. В эксперименте использовалось также удаление большей части поджелудочной железы (Меринг и Минковский), выключение В-клеток с помощью антител к ним или инсулину, избыточное введение контринсулярных гормонов.
К факторам риска следует отнести и чрезмерное употребление в пищу углеводов и жиров, что может со временем привести к перенапряжению и истощению функций В- клеток островков Лангерганса, а также психические травмы (например, сахарный диабет «биржевиков»). Отмечена зависимость сахарного диабета от массы тела новорожденного
– чем выше вес младенца, тем чаще в постнатальном периоде развивается сахарный диабет. Вскармливание грудных детей не кипяченым коровьим молоком, содержащим сывороточный бычий альбумин (фактор СБА) также ведет к сахарному диабету. Тем не менее, многие из вышеперечисленных факторов действуют на фоне наследственной предрасположенности, связь которой весьма высока (до 50% для ИЗСД и до 100% ИНСД).
Патогенез сахарного диабета. Поскольку существует два типа сахарного диабета – ИЗСД и ИНСД следует рассматривать патогенез для каждого из них отдельно. ИНСД – это семейное заболевание, где фактором риска, влияющим на формирование заболевания, является ожирение, но в отличие от ИЗСД здесь нет генетических маркеров. Патогенез ИНСД связывают с тремя механизмами, выявляющимися в той или иной степени у каждого больного:
1) недостаточная секреция инсулина для утилизации глюкозы, особенно в течение первого часа после ее поступления из-за снижения чувствительности глюкорецепторов В- клеток островков к глюкозо-стимулу;
2)инсулинорезистентность вследствие аномалий молекулы инсулина, связывания инсулина циркулирующими в крови антителами к нему, наличием антител к инсулиновым рецепторам в клетках, уменьшением их числа и т.п.;
3)усиленным образованием глюкозы печенью на протяжении полных суток, тогда как в норме синтез глюкозы происходит только днем.
Патогенез ИЗСД, так или иначе, связан с деструкцией и снижением числа активных β-клеток, резким уменьшением количества вырабатываемого инсулина, что приводит к абсолютному его дефициту. Хорошо известно, что по мере развития болезни до клинически выраженных форм диабета масса поджелудочной железы может уменьшиться
вдва раза, вес островков – в 3,3 раза, а количество В-клеток – в 850 (!) раз. В результате
дефицита инсулина нарушаются углеводный, жировой, белковый, водно-минеральный обмен, а также кислотно-щелочное равновесие.
Нарушение углеводного обмена при сахарном диабете связано, в первую очередь, с угнетением активности фермента гексокиназы в инсулинзависимых тканях – печени, мышцах, жировой и соединительной ткани, а также нарушением проницаемости клеточных мембран. Дефицит глюкозо-6-фосфата в печени компенсируется образованием его в процессе глюконеогенеза, а повышение активности фосфорилазы и глюкозо-6- фосфатазы печени способствует усиленному образованию глюкозы и тормозит синтез гликогена. В свою очередь, из-за повышенного уровня в крови контринсулярных гормонов, в частности глюкокортикоидов, активируются глюконеогенез и гликогенолиз, что ведет к гипергликемии. Таким образом, связанная с дефицитом инсулина гипергликемия обусловлена несколькими факторами:
1)затруднением перехода глюкозы из плазмы в клетку через транспортеры – глюты, особенно глют-4, локализованные в цитоплазматических мембранах и дальнейшая
ееутилизация (особенно инсулин зависимыми тканями – мышцами, жировой тканью, где преимущественно находятся эти траспортеры);
2)сниженной активностью гексокиназы клеток и ограничением вследствие этого превращения глюкозы по всем известным метаболическим путям;
3)активацией процессов распада гликогена (гликогенолиз);
4)образованием глюкозы из аминокислот, глицерина и жирных кислот и продуктов распада углеводов – лактата и пирувата (глюконеогенез);
5)торможением образования гликогена из глюкозы (гликогенез);
6)усиленной продукцией глюкозы печенью (так, у здорового человека ночью глюкоза печенью не вырабатывается, а у больных синтез глюкозы идет в течение суток постоянно, причем ночью он усилен, особенно ближе к утренним часам);
7)ослабленным липогенезом и усиленным липолизом с последующим превращением жиров в сахара;
8)угнетением пентозофосфатного пути превращения глюкозы и образования
НАДФ+.
К тому же усиливается превращение глюкозы по альтернативным инсулин независимым путям, например, сорбитоловому, в результате чего в организме накапливаются соответствующие продукты распада глюкозы: сорбитол и фруктоза, которые вследствие избыточного образования могут откладываться в таких тканях, как хрусталик, эндотелий сосудов, нервной системе.
Нарушения перехода и последующего превращения глюкозы в тканях приводит не только к гипергликемии, но и гиперлактацидемии – повышенному содержанию молочной кислоты в крови (норма 1,1-1,2 ммоль/л). Образовавшаяся вследствие нарушения аэробного окисления глюкозы молочная кислота, особенно в больших количествах, поступает в кровь из работающих мышц, селезенки, почек и легких и не успевает ресинтезироваться в гликоген печенью.
Инсулиновая недостаточность, гипергликемия и последующее изменение активности ферментов в почках (угнетение активности гексокиназы, повышение активности фосфатазы) нарушают нормальные процессы обратного всасывания глюкозы из первичной мочи, концентрация которой прогрессивно увеличивается по мере нарастания гипергликемии. Гипергликемия сопровождается повышением осмотического давления крови и переходом жидкости в кровь из тканей, что ведет к дегидратации последних. В результате обезвоживания тканей, гиперосмолярности крови возникает жажда – полидипсия. Определенный вклад в развитие полидипсии вносит полиурия.
Сам по себе патогенез полиурии связан, во-первых, с глюкозурией (содержащаяся в конечной моче глюкоза удерживает около себя воду), во-вторых, с дополнительным выведением с мочой электролитов (Na+ и K+, так называемый осмотический диурез), аминокислот и кетоновых тел. Кроме того, важным звеном патогенеза полиурии является
полидипсия.
Нарушение жирового обмена. Чаще всего нарушение жирового обмена происходит вторично вследствие первичного расстройства углеводного обмена. Проявлениями нарушения жирового обмена являются:
1)гиперлипемия (содержание липидов в плазме выше 8 г/л, норма 4-8);
2)гиперкетонемия (содержание кетоновых тел в плазме выше 30 мг/л);
3)гиперхолестеринемия (более 7,5 ммоль/л, норма 4,2-7,5);
4)гиперфосфолипидемия (более 3,5 ммоль/л, норма 2,0-3,5);
5)повышение содержания неэстерифицированных жирных кислот (более 0,8
ммоль/л);
6)увеличение содержания триглицеридов – триглицеридемия (более 1,9 ммоль/л,
норма 0,1-1,9);
7)увеличение содержания липопротеидов (более 8,6 г/л, норма 3,8-8,6). Перечисленные показатели измененного жирового обмена при сахарном диабете
обусловлены не только дефицитом инсулина, но и избытком контринсулярных гормонов, а также отсутствием липокаина. Гиперлипемия в отсутствии липокаина может приводить
кжировой инфильтрации печени, чему способствуют
1)обеднение печени гликогеном;
2)дефицит липотропных факторов, включая липокаин;
3)жировая диета;
4)избыток СТГ;
5)инфекции и интоксикации.
Эти же факторы приводят к кетозу, однако, непосредственными причинами кетоза являются следующие:
1)усиленный распад неэстерифицированных жирных кислот в печени;
2)нарушение ресинтеза ацетоуксусной кислоты в высшие жирные кислоты;
3)недостаточное окисление ацетоуксусной кислоты в цикле Кребса;
4)повышенное образование ацетоуксусной кислоты в печени.
Вышеописанные изменения жирового обмена ведут к ускорению развития атеросклероза. Нарушение белкового обмена. Эти нарушения касаются усиленного распада
протеидов и ослабления синтеза белка. Торможение синтеза белка является предпосылкой образования из их компонентов углеводов – глюконеогенез, который стимулируется под влиянием глюкокортикоидов и АКТГ. Нарушается белковый состав плазмы:
1)снижается содержание альбуминов,
2)растет концентрация глобулинов,
3)повышается уровень альфа-2-гликопротеидов.
Усиление глюконеогенеза сопровождается повышенным образованием свободных аминокислот, аммиака, мочевины и других продуктов распада белка, что ведет к гиперазотемии и гиперазотурии. Гипергликемия вызывает гликозилирование различных белков (например, альбумина, гемоглобина, белков базальной мембраны сосудов и т.д.), повышение иммуногенности, что играет важную роль в патогенезе микроангиопатий. Так, в крови здоровых людей содержится несколько разновидностей гемоглобина А (Hb1A, Hb1B, Hb1C), которые на конце -цепи содержат глюкозу или глюкозо-6-фосфат. В норме содержание таких гликозилированных гемоглобинов колеблется в пределах 4-6%, а у больных сахарным диабетом – 10-15%.
Концентрация гликозилированных гемоглобинов соответствует не уровню глюкозы в крови в момент взятия пробы, а его усредненному значению за предшествующий 4-6 недельный период. Нарушение белкового обмена сопровождается снижением продукции защитных белков, что объясняет склонность больных сахарным диабетом к привходящей инфекции. Наиболее частыми осложнениями сахарного диабета являются:
1) трофические кожные расстройства;
2)интеркуррентные инфекции;
3)сосудистые расстройства – атеросклероз, инфаркты миокарда, инсульты и
другие,
4)диабетические ретинопатия,
5)нефропатия,
6)нейропатия.
Наиболее грозным осложнением сахарного диабета, представляющим угрозу жизни, является кома. Для сахарного диабета характерны три типа комы:
1)кетоацидотическая,
2)гиперлактацидемическая,
3)гиперосмолярная.
Для купирования коматозного состояния используют введение инсулина, передозировка которого может вызвать гипогликемическое состояние с последующим переходом в гипогликемическую кому. В патогенезе кетоацидотической комы имеют значение следующие факторы:
1)дефицит инсулина,
2)гиперпродукция контринсулярных гормонов, особенно глюкагона,
3)прогрессирующая гипергликемия (из-за глюконеогенеза, гликогенолиза, нарушения утилизации глюкозы),
4)гиперосмолярность крови (что вызывает переход жидкости из тканей в кровь и формирует дегидратацию тканей),
5)полиурия с высокой осмолярностью мочи (что ведет к потере жидкости, электролитов, гиповолемии с последующим снижением артериального давления),
6)прогрессирующее нарушение жирового обмена (гиперлипидемия, кетоз),
7)кетоз сменяется кетоацидозом и далее метаболическим ацидозом. Таким образом, кетоацидотическая кома является результатом:
1)отравления организма и ЦНС кетоновыми телами,
2)обезвоживания,
3)метаболического ацидоза.
В патогенезе гиперосмолярной комы, которая встречается в 10 раз реже кетоацидотической, имеют значения следующие факторы:
1)дефицит инсулина,
2)резко выраженная гипергликемия (до 55,5-111 ммоль/л),
3)гиперосмолярность крови (за счет гипергликемии и гипернатриемии),
4)дегидратация тканей,
5)отсутствие кетоацидоза,
6)гиповолемия,
7)осмотический диурез и прогрессирующая потеря электролитов.
Впатогенезе гиперлактацидемической комы имеют значение следующие факторы:
1)дефицит инсулина,
2)гиперпродукция контринсулярных гормонов (СТГ, глюкагона и других),
3)накопление пировиноградной кислоты и переход ее в молочную – гиперлактацидемия,
4)невысокая гипергликемия,
5)метаболический ацидоз за счет гиперлактацидемии,
6)отсутствие гиперкетонемии и кетонурии.
Гипогликемическая кома. В патогенезе гипогликемической комы имеет значение предшествующее гипогликемическое состояние (содержание глюкозы в крови ниже 4,44 ммоль/л) с характерной симптоматикой. Важное значение приобретает передозировка инсулина, ведущая к гипогликемии (ниже 2,22 ммоль/л); гипогликемия приводит к нарушению энергетического обмена мозга и, в первую очередь, нейронов коры больших полушарий. В дальнейшем появляются функциональные нарушения ЦНС, а также
развиваются компенсаторные реакции в виде активации симпатоадреналовой системы, выброса АКТГ и других контринсулярных гормонов, развития судорог и других. Немаловажное значение в патогенезе приобретают падение осмотического давления крови, а также спазм сосудов головного мозга.
Патофизиология нарушения гормональной регуляции водно-солевого обмена
Специальная группа гормонов отвечает за осморегуляцию (уровень осмотического давления крови) и поддержание определенного соотношения ионов натрия и калия. Главные гормоны этой группы – вазопрессин и альдостерон.
При заболевании гипоталамо-нейрогипофизарной системы, связанном с недостаточностью продукции АДГ, развивается несахарный диабет.
Несахарный диабет – это заболевание, характеризующееся полидипсией и полиурией с низкой относительной плотностью мочи.
Этиологические факторы несахарного диабета весьма разнообразны: заболеванию могут предшествовать травмы черепа, многочисленные инфекции (малярия, скарлатина, бруцеллез и др.). Особенно часто развитие несахарного диабета связывается с перенесенным гриппом.
Внорме АДГ действует в дистальных отделах почечных канальцев, активируя реабсорбцию воды, уменьшая диурез. При недостаточности АДГ уменьшается концентрационная способность почек, увеличивается диурез, снижается относительная плотность мочи. Большая потеря жидкости и изменение физико-химического состава крови приводят к раздражению «питьевого» центра и компенсаторной жажде, обеспечивая пополнение водных ресурсов организма. Величина суточного диуреза резко возрастает (до 30 л мочи в сутки).
В1955 г. Конн впервые описал клинику синдрома, при котором наблюдаются перемежающая тетания, периодическая сильная мышечная слабость, парестезии, полиурия и полидипсия, гипертензия и нарушение функции почек. Ведущую роль в возникновении синдрома играет повышенная секреция альдостерона клубочковой зоной коры надпочечников. В 85% случаев причиной этого заболевания является наличие гормонально активной опухоли клубочковой зоны – альдостеромы.
Синдром Конна (первичный альдостеронизм) – относительно редкое заболевание, встречающееся преимущественно у женщин, чаще в возрасте 30-45 лет. При длительном повышенном выделении альдостерона развиваются перечисленные выше симптомы, связанные с изменением количества и нарушением соотношения электролитов в организме. Условно симптомы первичного альдостеронизма могут быть разделены на три группы:
1 нервно-мышечные (тетания, мышечная слабость, парестезии, спазмы мышц, судороги);
2 почечные (полиурия, полидипсия, ночное мочеиспускание, никтурия); 3 связанные с гипертензией (ослабление зрения, головные боли, головокружение).
Основным и первичным проявлением воздействия избытка альдостерона при синдроме Конна является развитие гипокалиемии в связи с потерей калия с мочой.
Взамен потерянного калия, вышедшего из клеток во внеклеточную жидкость для восполнения соответствующих потерь, внутрь клеток поступают ионы натрия, хлора и водорода. Это ведет к развитию внутриклеточного ацидоза, а потеря калия с мочой и поступление хлора внутрь клеток – к гипокалиемическому, гипохлоремическому алкалозу, что обусловливает развитие тетании при нормальной концентрации кальция и фосфора в крови. Длительная гиперкалийурия ведет к дегенерации эпителия почечных канальцев, в связи с чем уменьшается чувствительность последних к АДГ. В результате появляется полиурия, которая сопровождается выраженной жаждой (полидипсией). Полиурией объясняется редкость возникновения отеков при синдроме Конна. При потере значительного количества калия (10-30%) отмечаются мышечная слабость, апатия, потеря аппетита.
Для первичного альдостеронизма характерно возникновение артериальной гипертонии (протекающей иногда в тяжелой форме), которая объясняется повышением тонуса артериол. Последнее связано с перераспределением электролитов и задержкой натрия в сосудистой стенке, с последующим набуханием стенки сосуда и сужением просвета сосуда. К тому же задержка натрия в интрацеллюлярной жидкости повышает реактивность симпатической нервной системы в связи с потенцирующим действием норадреналина. У больных обнаруживается усиленное выделение с мочой альдостерона.
Ряд неэндокринных заболеваний (гипертоническая болезнь, заболевание почек, сердечная недостаточность различного генеза, цирроз печени и т.д.) может сопровождаться так называемым вторичным альдостеронизмом.
Повышенное выделение альдостерона у больных гипертонической болезнью происходит тогда, когда в патогенетическую цепь включается почечное звено: при развитии ишемии почек происходит выделение юкстагломерулярным аппаратом ренина, который способствует синтезу ангиотензина-I. Образующийся из него ангиотезин-II стимулирует секрецию альдостерона. Повышенная секреция альдостерона влечет за собой изменение соотношения в тканях натрия и калия, а это потенцирует эффекты катехоламинов, усиливает тонус и набухание гладкомышечных волокон артериол и, таким образом, способствует прогрессированию артериальной гипертензии.
При сердечной недостаточности, нефрозах, циррозах печени вторичный альдостеронизм имеет определенное значение в патогенезе развития отеков, однако не является его первопричиной, представляя собой лишь дополнительный фактор в связи с вызываемым им усилением задержки натрия. Задержка натрия в тканях при альдостеронизме только тогда может сопровождаться отеками, когда имеет место предотечное состояние.
Патофизиология нарушения гормональной регуляции обмена кальция и фосфора
Организм защищает себя от гипокальциемии, увеличивая абсорбцию кальция в пищеварительном тракте, уменьшая почечную экскрецию и повышая скорость разрушения костей и деминерализации. И наоборот: высокие концентрации кальция во внеклеточном пространстве приводят к снижению его абсорбции в пищеварительном тракте, увеличению экскреции почками и усилению минерализации костей.
Околощитовидными железами вырабатывается паратиреоидный гормон (паратгормон), под влиянием которого концентрация кальция в крови повышается (гиперкальциемический эффект).
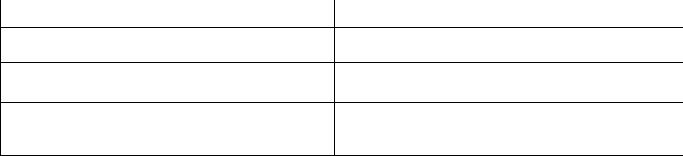
Адекватным регулятором функции околощитовидных желез служит изменение уровня кальция сыворотки: понижение его активирует, а увеличение тормозит секрецию паратиреоидного гормона кальцитонина (Таблица 5).
В щитовидной железе одновременно продуцируется тирокальцитонин (антагонист паратгормона), который, напротив, способствует снижению кальциемии (гипокальциемический фактор). В норме паратгормон и тирокальцитонин находятся в динамическом равновесии.
Наряду с регуляцией кальциевого метаболизма околощитовидные железы оказывают сильное влияние на обмен фосфора, снижая его концентрацию в крови.
Гипопаратироз. Заболевание развивается как следствие недостаточности функции околощитовидных желез. Уменьшение продукции паратгормона может обусловливаться удалением части околощитовидных желез, ослаблением их деятельности при инфекциях и интоксикациях.
Различают острую и хроническую паратиреоидную недостаточность.
Причиной возникновения острой паратиреоидной недостаточности (паратиропривной тетании) чаще всего является случайное удаление околощитовидных желез при операциях на щитовидной железе или травматическое их повреждение. Уже в первые сутки развивается депрессивное состояние, возбудимость нервов и мышц
возрастает, затем начинаются подергивания отдельных мышц головы и туловища, постепенно переходящие в тонические судороги.
Содержание кальция в крови во время тетанического припадка оказывается резко сниженным. При снижении концентрации кальция в крови нарушается равновесие между моновалентными и двухвалентными катионами (натрия и калия к кальцию и магнию), в результате чего и повышается нервно-мышечная возбудимость.
Табли
|
ца 5 |
Механизм действия паратгормона и |
|
|
|
Паратгормон |
Кальцитонин |
Вкостях стимулирует мобилизацию и выход в В костях стимулирует отложение Са2+ кровь Са2+
Вкишечнике усиливает всасывание Са2+ в В кишечнике тормозит всасывание Са2+ и фосфато
кровь
В почках усиливает реабсорбцию Са2+ и В почках усиливает экскрецию Са2+ тормозит реабсорбцию фосфатных ионов
Содержание калия в крови больных гипопаратирозом нередко повышено, содержание фосфора в крови во время приступа также значительно увеличивается. При тетании наблюдается сдвиг рН крови в кислую сторону. Удаление всех паращитовидных желез всегда заканчивается летальным исходом.
Хроническая форма околощитовидной недостаточности характеризуется подавлением аппетита, прогрессирующей кахексией и рядом трофических расстройств (гиперемия, кровоизлияния в области привратника желудка и двенадцатиперстной кишки с образованием язв, выпадение волос, в молодом возрасте – замедление роста).
Псевдогипопаратироз (болезнь Олбрайта). В отличие от истинного гипопаратироза, обусловленного первичной недостаточностью околощитовидных желез, является врожденным заболеванием. Характеризуется нанизмом (низким ростом), аномалиями в развитии скелета, дистрофическими изменениями зубов и костей и отставанием в интеллектуальном развитии. Наблюдаются снижение содержания кальция и повышение содержания фосфора в крови, гипофосфатурия. В противоположность истинному гипопаратирозу отмечается резистентность к паратгормону.
Гиперпаратироз (генерализованная фиброзная остеодистрофия, болезнь Реклингаузена). В 1876 году английский врач Педжет описал случай заболевания, проявившегося резкой деформацией костей. На секции кости оказались утолщенными и вместе с тем легкими, порозными и настолько мягкими, что их можно было резать ножом.
Гиперпаратироз – сложный и многообразный симптомокомплекс, клинические проявления которого обусловлены избыточной продукцией паратиреоидного гормона.
Патологические изменения при гиперпаратирозе происходят, главным образом, в костях и почках. Костная ткань подвергается рассасыванию вследствие усиленной остеокластической реакции и деминерализации.
Нормальное пластинчатое костное вещество замещается свободной от извести молодой остеоидной тканью. Кости делаются мягкими, происходят разнообразные, иногда причудливые, деформации скелета.
Отмечаются резкие изменения в минеральном обмене. Паратироидный гормон понижает почечный порог в отношении фосфора, ослабляя его реабсорбцию в нефронах и усиливая экскрецию фосфатов с мочой.
При гиперпаратирозе потеря фосфора приводит к мобилизации его неорганических соединений из костей. Поскольку эти соединения являются солями кальция, последний,
освобождаясь из костей, в избытке поступает в кровь и развивается гиперкальциемия. Кроме того, паратгормон стимулирует всасывание кальция из кишечника,
функционально перекрываясь с витамином D, а также обратное всасывание кальция из клубочкового фильтрата.
Различают первичный и вторичный гиперпаратироз.
Первичный гиперпаратироз. Обусловлен развитием аденомы или гиперплазией околощитовидных желез.
Вторичный гиперпаратироз. Гиперплазия и гиперфункция околощитовидных желез развиваются в результате первичного изменения кальциево-фосфорного обмена (потери кальция и накопления фосфора в крови).
Вторичный гиперпаратироз имеет компенсаторный характер. Чаще всего он возникает при почечной остеодистрофии, когда нарушается экскреция фосфорнокислых солей в результате клубочковой недостаточности или же вследствие нарушения обмена ионов в дистальных канальцах.
Патофизиология нарушения гормональной регуляции артериального давления
Уровень артериального давления контролируется и поддерживается нейрогуморальными факторами при участии определенных вазомоторных центров, расположенных в продолговатом мозге.
В регуляции тонуса сосудов принимают участие и многие гормоны:
1глюкокортикоидные гормоны коры надпочечников;
2минералокортикоидные гормоны коры надпочечников (выработка и выделение альдостерона в норме стимулируются ионами калия и ангиотензином; минералокортикоиды увеличивают резорбцию натрия в дистальной части нефрона; если они присутствуют в избыточном или неконтролируемом количестве, повышение общего содержания натрия в организме ведет к увеличению объема жидкости, возрастанию объема крови и подъему артериального давления; кроме того, прямое воздействие минералокортикоидов на гладкие мышцы повышает сопротивление периферических сосудов);
3катехоламины, вырабатываемые мозговым веществом надпочечников (оказывают разнообразное влияние на функцию сердечно-сосудистой системы и метаболизм, обеспечивая доставку крови и питательных веществ в условиях выраженного стресса);
4ренин-ангиотензин;
5вазопрессин.
Действие гормонов, вовлеченных в регуляцию артериального давления, наиболее отчетливо проявляется при их избытке или недостатке.
Избыточная продукция корой надпочечников гормона роста, глюкокортикоидных и особенно минералокортикоидных гормонов, катехоламинов, гипертироз, гиперпаратироз, повышенная продукция ренин-ангиотензина могут вызвать значительное повышение артериального давления.
Недостаточность надпочечников (болезнь Аддисона), гипотиреоз, автономная диабетическая нейропатия провоцируют возникновение гипотензии. При низком содержании кортизола в плазме нарушается реактивность гладких мышц сосудов, что вносит определенный вклад в развитие гипотензии. Механизм последнего при адреналовой недостаточности отчасти связан с уменьшением объема жидкости, обусловленным дефицитом альдостерона.
Артериальная гипертензия может быть проявлением гипертонической болезни (эссенциальная гипертензия, первичная артериальная гипертензия).
Термин «эссенциальная гипертензия» используется для описания состояния пациентов, у которых неизвестна причина повышения артериального давления. У большинства больных с гипертензией отсутствуют какие-либо симптомы, а данные лабораторных исследований остаются в пределах нормальных значений. При обследовании обычно выявляется высокое артериальное давление, а иногда и связанные с