

Прогноз и профилактика
Прогрессирующая мышечная дистрофия Беккера имеет неблагоприятный прогноз. Хотя обездвиженность у пациентов наступает гораздо позже, чем при дистрофии Дюшенна, в конечном итоге поражение сердечной мышцы и дыхательной мускулатуры приводят к гибели пациентов от сердечной или дыхательной недостаточности. Продуманный уход, адекватная терапия, вентиляционная поддержка дыхания, применение ортопедических средств могут лишь увеличить продолжительность и улучшить качество жизни пациента.
Профилактика заключается в предупреждении рождения ребенка с патологией путем генетического консультирования будущих родителей и проведение пренатальной диагностики.
63. Конечностно-поясная прогрессирующая мышечная дистрофия, лопаточно-бедренный тип Эрба (тип II а). Этиология, патоморфология, клиника, диагностика, лечение, профилактика.
В настоящее время описано 2 генетических типа данного заболевания и 10 подтипов. Из них подтипы 1А и 1В имеют аутосомно-доминантную передачу с неизвестным пока характером биохимического дефекта и типа мутаций гена. Подтипы 2А передаются по аутосомно-рецессивному типу, для большинства из них установлен характер мутации и локализация патологического гена.
Конечностно-поясная мышечная дистрофия 2А типа (КПМД2А) – аутосомно-рецессивное наследственное заболевание, входящее в гетерогенную группу клинически полиморфных генетических мышечных болезней с преимущественным поражением мышц тазового и плечевого поясов.
Этиология и патогенез
За развитие заболевания ответственен ген CAPN3, находящийся на 15-й хромосоме и содержащий 24 экзона. Нормальным продуктом гена CAPN3 является белок кальпаин-3 – представитель широкого семейства цитозольных Са2+-активируемых цистеиновых протеаз, ответственных за селективную деградацию белков.
Поскольку нет четкого представления о всех функциях белка кальпаина-3, точные патогенетические механизмы развития КПМД2А до сих пор неизвестны. Показаны ключевая роль активации окислительного стресса (рост токсичных реактивных форм кислорода) и вторичное нарушение убиквитинопосредованной деградации белка.
Патоморфология
Характеризуется перерождением мышечной ткани, замещением ее жировой и соединительной тканью, некрозом отдельных волокон.
Клинические проявления
Первые признаки заболевания при аутосомно-рецессивном варианте проявляются преимущественно в 14-16 лет, реже - в 5-10-летнем возрасте.
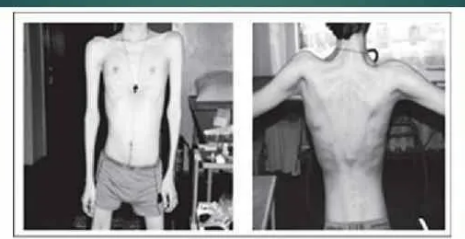
Кальпаинопатия (КПМД2А) характеризуется симметричной прогрессирующей слабостью проксимальных групп мышц – плечевого и тазового поясов. Характерными симптомами являются мышечная слабость, патологическая мышечная утомляемость при физической нагрузке, изменение походки по типу «утиной» или «генеральской». В начале болезни атрофии локализуются в проксимальных группах мышц нижних конечностей, в более поздних стадиях в процесс вовлекаются мышцы спины и живота, дистальных отделов конечностей. Иногда миодистрофический процесс одновременно поражает мышцы тазового и плечевого пояса. Вследствие атрофий возникают гиперлордоз, «крыловидные» лопатки, «осиная» талия, «пустые» надплечья. При вставании больные применяют вспомогательные приемы. Псевдогипертрофии мышц, контрактуры суставов, сухожильные ретракции, как правило, выражены умеренно. Кардиомиопатии, как правило, редко развиваются; интеллект сохранен.
Миодистрофия Эрба-Рота сопровождается ранним угасанием коленных рефлексов, а также сухожильных рефлексов с плечевого бицепса и трицепса. Чувствительная сфера не нарушена. Со временем атрофия и слабость дыхательных мышц приводит к появлению прогрессирующей дыхательной недостаточности, возникает опасность развития застойной пневмонии. Миодистрофический процесс в гладкой мускулатуре обуславливает снижение кишечной перистальтики с тенденцией к запорам. Поражение сердечной мышцы влечет за собой возникновение кардиомиопатии, нарушений ритма, сердечной недостаточности.
Диагностика и дифференциальный диагноз
Вследствие генетической гетерогенности этой группы ПМД основным при постановке диагноза является проведение ДНК анализа, что позволяет установить форму заболевания и определить прогноз. Отрицательный анализ ДНК является показанием к биопсии мышц. В биоптате обнаруживаются различные по толщине мышечные волокна, уменьшенное число мышечных ядер, некротические и склеротические изменения.
При неврологическом осмотре обращает на себя внимание снижение мышечной силы в мускулатуре проксимальных отделов ног и рук, гипотония и гипотрофия указанных мышц, гипорефлексия или полное выпадение локтевых и коленных рефлексов, сохранность всех видов чувствительности. ЭМГ и ЭНГ свидетельствуют о первичном поражении мышечной ткани при сохранности проведения импульсов по нервным стволам. Проведение электромиографии, как стимуляционной, так и игольчатой, позволяет исключить нейрональный и невральный уровни поражения, заподозрить первично-мышечную заинтересованность, а последующее комплексное лабораторное обследование дает возможность уточнить генез выявленной патологии.
Резкое повышение уровня креатинфосфокиназы характерно для начального периода дистрофии Эрба-Рота, затем происходит постепенное понижение этого показателя вплоть до нормальных цифр.
МРТ всего тела позволяет оценить распространенность, симметричность и выраженность мышечной патологии (в частности, степень замещения мышц жировой тканью), а также определить избирательное вовлечение тех или иных групп мышц туловища и конечностей, что в конечном итоге облегчает поиск мутантного гена в ходе дальнейшего генетического обследования.
Таким образом, существует перечень симптомов, позволяющих заподозрить кальпаинопатию при наличии клиники прогрессирующей КПМД:
наличие проксимального вялого тетрапареза с преимущественным вовлечением мышц задних отделов нижних конечностей, приводящих мышц бедра и двуглавых мышц плеча;
отсутствие вовлечения мышц лица, языка и шеи;
отсутствие признаков кардиомиопатии и интеллектуальных нарушений;
начало в любом возрасте после периода нормального физического развития; значительное повышение концентрации сывороточной KФК;
наличие данных, подтверждающих аутосомно-рецессивный тип наследования.
Лечение
Основная цель терапии при любой форме прогрессирующих наследственных мышечных дистрофий, в том числе и при КПМД2А, заключается в улучшении качества жизни пациента и продлении ее активной фазы
Медикаментозная терапия
Препараты, улучшающие тканевой метаболизм (АТФ, коэнзимQ, рибоксин, актовегин, кортексин).
Аминокислотные комплексы (церебролизин, глицин, метионин, глутаминовая, фолиевая кислоты).
Физиотерапия (обертывания и электрофорез с ферментными препаратами)
Лечебная физкультура
Ортопедическая коррекция. Для профилактики остеопороза показано назначение препаратов, содержащих витамин D3 и кальций.
При поражении сердечной мышцы рекомендован инозин, сердечные гликозиды, антиаритмики. При развитии контрактур может потребоваться ортопедическое лечение. Выраженное снижение жизненной емкости легких из-за атрофии дыхательных мышц служит показанием к ИВЛ.