

89. Болезнь Марфана. Этиология, патоморфология, патогенез, клиника, диагностика, лечение, профилактика.
Синдромом Марфана (СМ), или Марфана-Ашара, называют аутосомно-доминантное наследственное заболевание соединительной ткани с преимущественным поражением сердечно-сосудистой системы, скелета и органа зрения. Частота СМ в популяции составляет примерно 1:10000.
Этиология и патогенез
Синдром Марфана относится к врожденным аномалиям, наследуемым по аутосомно-доминантному типу, с выраженным плейотропизмом, варьирующей экспрессивностью и высокой пенетрантностью.
В основе синдрома Марфана лежат мутации в гене FBN1, отвечающем за синтез фибриллина – важнейшего структурного белка межклеточного матрикса, придающего эластичность и сократимость соединительной ткани. Аномалия и дефицит фибриллина при синдроме Марфана приводят к нарушению формирования волокнистых структур, потере прочности и упругости соединительной ткани, невозможности выдерживать физиологические нагрузки. Гистологическим изменениям в большей степени подвержены стенки сосудов эластического типа и связочный аппарат (в первую очередь, аорта и цинновая связка глаза, содержащие наибольшее количество фибриллина).
При генетическом исследовании в 75% случаев синдрома Марфана выявляется семейный тип наследования, в остальных - первичная мутация. Риск рождения ребенка с синдромом Марфана возрастает с увеличением возраста отца (особенно после 35 лет).
При болезни Марфана происходит замена нуклеотидов в гене, содержащем информацию о структуре пептида фибриллина-1. Этот белок относится к гликопротеидам, принимает участие в микрофибриллярном комплексе, он обеспечивает основу эластических фибрилл соединительной ткани. В крупных сосудах, связочном аппарате содержится большое количество эластиновых фибрилл, поражение которых и даёт основные клинические проявления синдрома.
Изменение структуры коллагеновых волокон приводит к нарушению первичного звена гемостаза. Эластические фибриллы имеют определенные механизмы участия в системе гемостаза - в сосудах происходит адгезия («прилипание») тромбоцитов к эластину через фибронектин. Регистрируется снижение его уровня в крови у людей с синдромом Марфана. Фибронектин, в свою очередь, образуется в клетках эндотелия и участвует в последующих репаративных процессах, создавая основу для производства других компонентов соединительной ткани — фибробластов. Таким образом, совершенно неоспоримо участие сосудистой стенки в реакциях свертываемости крови, и неизбежен вывод о возможных патологиях протекания нормальных гемостатических процессов при изменении состояния её структурных компонентов и процессов сосудистой регуляции.
Патоморфология
Обычная находка при СМ – тяжелые изменения со стороны аорты - дилатация (аннулоаортальная эктазия), аневризмы, расслоения, разрывы - и клапанов - расширение клапанных отверстий, патологическое удлинение хорд, их разрывы. Типичные для СМ гистологические изменения, называемые эрдгеймовским некрозом (кистозной медиальной дегенерацией), выявляются в средней оболочке сосудов эластического типа. Они характеризуются разрушением эластического каркаса с некрозом и фрагментацией эластических волокон, нарушением ориентации и расщеплением коллагеновых волокон, дистрофией гладкомышечных клеток, накоплением между волокнистыми структурами базофильного слизеподобного вещества (мукополисахарида) с формированием небольших кист.
Клиника
Скелетные аномалии включают:
высокий рост
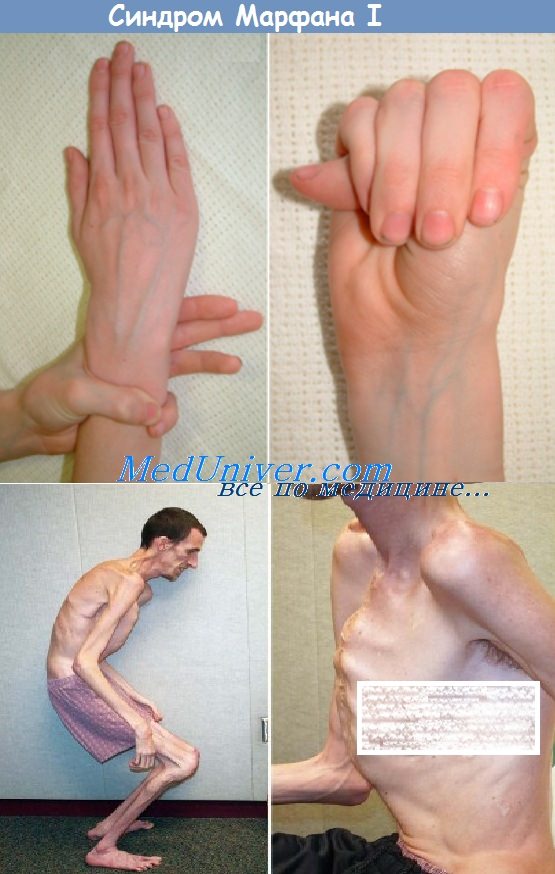
относительно короткое туловище с непропорционально длинными тонкими конечностями (долихостеномелией) и удлиненными паукообразными пальцами (арахнодактилией)
астеническое телосложение со слаборазвитой подкожной клетчаткой и мышечной гипотонией
длинный и узкий лицевой скелет (долихоцефалией)
наличие высокого аркообразного неба и нарушения прикуса (прогнатии)
гипермобильность суставов
деформация грудной клетки (воронкообразная или килевидная форма)
деформация позвоночника (сколиоз, кифоз, кифосколиоз, подвывихи и вывихи шейного отдела, спондилолистез), а также плоскостопие и протрузия вертлужной впадины.
Аномалии глаз включают подвывих хрусталиков, уплощение роговицы, удлинение глазного яблока и гипоплазию радужки.
Сердечнососудистые аномалии включают:
прогрессирующее расширение восходящей части и клапанного кольца аорты (дилатация, аннулоаортальная эктазия) и аневризмы;
поражение митрального клапана - дегенерация створок, патологическое удлинение и разрыв створочных хорд, обызвествление клапанного кольца
У плода возможно формирование врожденных пороков сердца - коарктации аорты, стеноза легочной артерии, ДМПП и ДМЖП.
Органические и функциональные изменения сердца и сосудов у больных синдромом Марфана часто сопровождаются нарушением ритма (наджелудочковой и желудочковой тахикардией, фибрилляцией предсердий) и развитием инфекционного эндокардита
При синдроме Марфана наблюдается поражение других систем и органов: нервной (эктазия твердой мозговой оболочки, в т. ч. пояснично-крестцовое менингоцеле), бронхолегочной (спонтанный пневмоторакс, эмфизема легких, дыхательная недостаточность), кожи и мягких тканей (атрофические стрии), рецидивирующие паховые и бедренные грыжи, вывихи и разрывы связок, а также эктопия почек, опущение мочевого пузыря и матки, варикозное расширение вен и другие.
Основными причинами гибели больных CМ являются разрыв раслаивающей аневризмы аорты и сердечная недостаточность.
Диагностика
Диагноз синдрома Марфана основывается на семейном анамнезе, наличии у больного типичных диагностических признаков по результатам физикального осмотра, ЭКГ и ЭхоКГ, офтальмологического и рентгенологического обследования, молекулярно-генетического анализа и лабораторных исследований.
ЭКГ при синдроме Марфана позволяет определить нарушение ритма сердца, выраженную гипертрофию миокарда; ЭхоКГ - обнаружить клапанную регургитацию, увеличение размеров левого желудочка, пролапс митрального клапана, разрывы хорд, дилатацию аорты. На рентгенографии грудной клетки можно увидеть расширение корня и дуги аорты, увеличение размеров сердца; на КТ и МРТ сердца и сосудов - выявить дилатацию и аневризмы аорты.
Аортография показана при подозрении на аневризму и расслоение аорты. Наличие эктопии хрусталика уточняют с помощью биомикроскопии и офтальмоскопии; протрузию вертлужной впадины устанавливают методом рентгенографии тазобедренных суставов; эктазию твердой мозговой оболочки – МРТ позвоночника.
При синдроме Марфана определяется возрастание (в 2 раза и более) почечной экскреции метаболитов соединительной ткани: глюкозоаминогликанов и их фракций. Метод прямого автоматического секвенирования ДНК позволяет провести генетическую идентификацию мутаций в гене FBN1.
Известны следующие фенотипические диагностические тесты СМ:
- соотношение кисть-рост > 11%;
- отношение размаха рук к росту > 1,05 - длина среднего пальца > 10 см;
- отношение длины верхнего сегмента тела к нижнему < 0,86;
- индекс телосложения Варги (ИВ) < 1,5 ИВ = масса тела, г/(рост, см)² - возраст, годы/100
Весьма часто при СМ бывают положительными тесты на арахнодактилию:
а) тест большого пальца Steinberg: согнутый 1-й палец выступает за мягкие ткани кисти. При рентгенографии кисти с приведенным большим пальцем его фаланга выступает за скелет метакарпальных костей;
б) тест запястья Walker-Murdoch: При обхватывании запястья одной руки другой. При арахнодактилии 1-й и 5-й пальцы соединяются друг с другом.
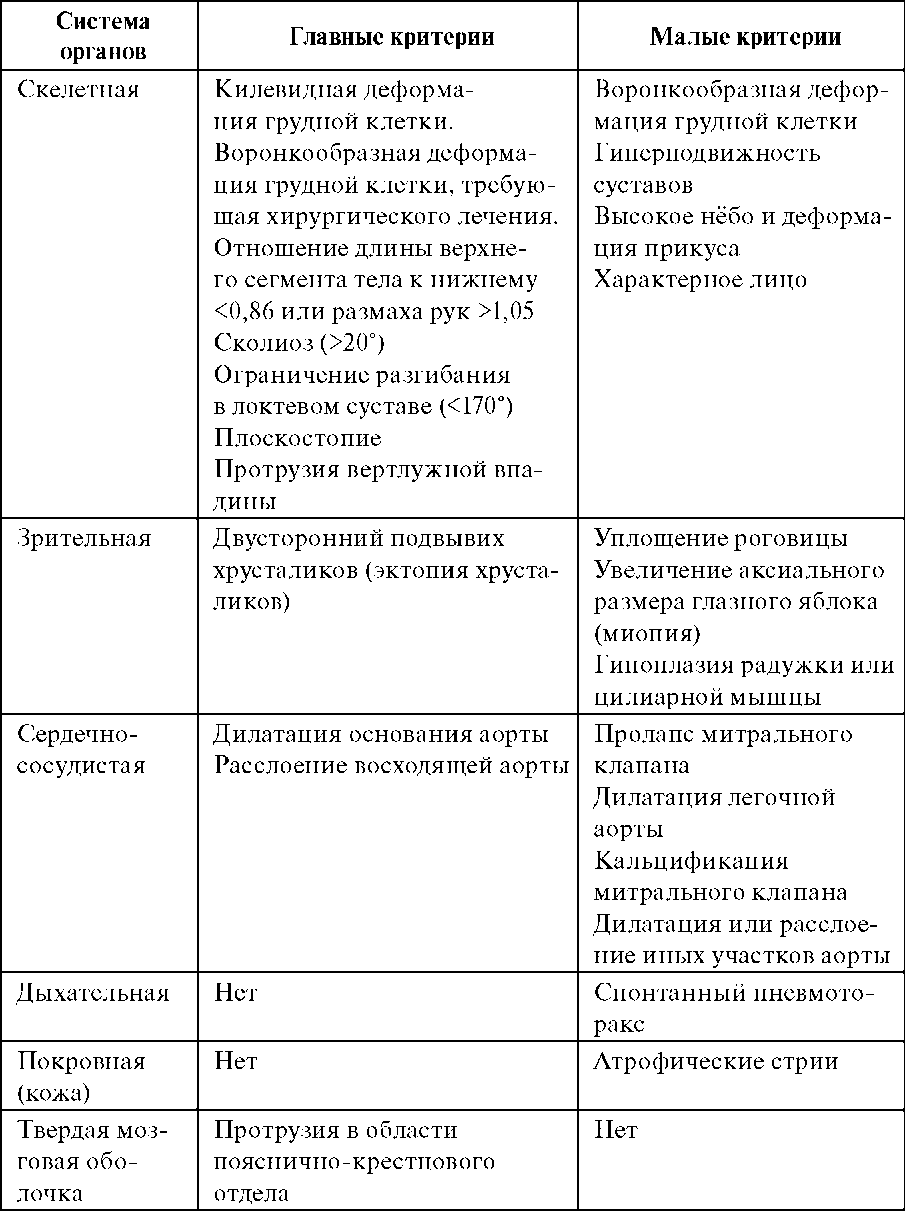
За диагностические критерии синдрома Марфана берутся характерные изменения в различных системах и органах.
Для установления диагноза синдрома Марфана необходимо наличие минимум по 1-му большому критерию в двух системах органов и 1-го малого - в третьей; в скелетной системе – присутствие минимум 4-х больших.
Лечение
Лечение и дальнейшее наблюдение пациентов с синдромом Марфана должно осуществляться группой специалистов: офтальмологом, кардиологом, кардиохирургом, ортопедом, генетиком, терапевтом.
Лечение больных с синдромом Марфана направлено на профилактику прогрессирования заболевания и развития осложнений, в первую очередь в сердечно-сосудистой системе. При диаметре аорты до 4 см назначаются β-адреноблокаторы, антагонисты кальция или ингибиторы АПФ. Хирургическое лечение проводится при недостаточности клапанов сердца, пролапсе митрального клапана, значительном расширении (>5 см) восходящей части и расслоении аорты.
Реконструктивные операции на аорте при синдроме Марфана, имеют высокий процент послеоперационной 5-ти и 10-ти летней выживаемости. При необходимости выполняют протезирование митрального клапана. У беременных с синдромом Марфана и выраженной сердечно-сосудистой патологией проводят досрочное оперативное родоразрешение путем кесарева сечения. С целью профилактика инфекционного эндокардита и тромбозов после операционных вмешательств назначаются антибиотики и антикоагулянты.
При синдроме Марфана проводится коррекция зрения с помощью подбора очков и контактных линз, при необходимости – лазерное или хирургическое лечение катаракты, глаукомы, удаление смещенного хрусталика с имплантацией искусственного. При выраженных скелетных нарушениях может потребоваться хирургическая стабилизация позвоночника, торакопластика, эндопротезирование тазобедренных суставов. Применяются также патогенетическая коллагеннормализующая терапия, метаболическая и витаминотерапия.
Прогноз
Прогноз жизни больных с синдромом Марфана определяется, в первую очередь, степенью сердечно-сосудистых изменений, а также поражений скелета и глаз. Имеется высокий риск осложненного течения, снижения продолжительности жизни (90-95% не доживают до 40-50 лет) и внезапной смерти. Своевременная кардиохирургическая коррекция при синдроме Марфана позволяет значительно увеличить продолжительность (до 60-70 лет) и улучшить качество жизни больных.
Больные синдромом Марфана должны находиться под постоянным врачебным наблюдением и регулярно проходить диагностическое обследование. При синдроме Марфана показан низкий или средний уровень физической активности. Женщинам детородного возраста с синдромом Марфана необходимо пройти медико-генетическое консультирование.
Классификация наследственных болезней с поражением нервной системы
I. Дегенеративные заболевания нервной системы