Лекции №25-26 Закономерности реакций нуклеофильного замещения
План.
Механизм реакций. Катализ. Соотношение между механизмами SN1 и SN2.
Влияние строения реагентов на реакции нуклеофильного замещения.
Конкуренция реакций нуклеофильного замещения. Правило Корнблюма.
Реакции некаталитического нуклеофильного замещения могут протекать в зависимости от условий, характера реакционной среды, структуры субстрата и природы нуклеофильного реагента по одностадийному или двухстадийному механизмам.
О дностадийный
процесс представляет собой элементарную
реакцию SN2-замещения,
где индекс 2 указывает на бимолекулярность
реакции:
дностадийный
процесс представляет собой элементарную
реакцию SN2-замещения,
где индекс 2 указывает на бимолекулярность
реакции:
или более схематично
:Y-
+ RX
![]()
RY + X:-
RY + X:-
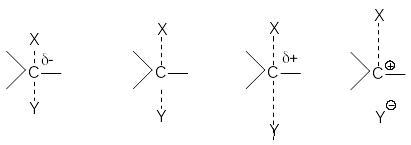
Можно видеть, что нуклеофил своей неподеленной парой электронов атакует углеродный атом в молекуле субстрата со стороны, противоположной замещаемой группе. При образовании активированного комплекса атакуемый атом углерода переходит из sp3-состояния в sp2-состояние. В результате углеродный атом оказывается в одной плоскости с группами R, R’ и R”, а нуклеофил и замещаемая группа связываются с ним за счет его р-орбитали. Распад активированного комплекса в продукт реакции сопровождается обратным переходом углеродного атома в sp3-состояние и обращением конфигурации исходного соединения, которое вызвано присоединением группы Y со стороны, противоположной разорвавшейся связи С-Х. Поэтому если объектом бимолекулярного нуклеофильного замещения является хиральный атом углерода, то в результате реакции происходит обращение оптической активности соединения. Скорость бимолекулярной реакции SN2 выражается уравнением
r = k[RX][Y-] (1)
При двухстадийном нуклеофильном замещении разрыв связи С-Х и образование связи С-Y происходит не одновременно. Первая элементарная стадия этой реакции - мономолекулярный гетеролитический разрыв связи С-Х с образованием карбкатиона

При этом центральный атом углерода переходит в состояние sp2-гибридизации и карбкатион приобретает плоское строение. Поэтому он с равной вероятностью атакуется нуклеофилом как со стороны отщепившейся группы, так и с противоположной стороны.
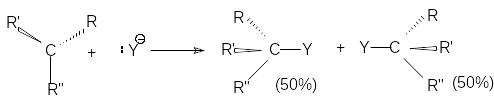
Поэтому, если объектом нуклеофильной атаки является хиральный атом углерода, то в результате реакции оптически активного субстрата образуется рацемическая смесь.
Первая стадия - мономолекулярная стадия гетеролиза - является скорость-определяющей. В связи с этим двухстадийный процесс нуклеофильного замещения обозначают символом SN1, где цифра 1 означает мономолекулярность скорость-определяющей стадии.
Скорость реакций SN1 в соответствии с характером скорость-определяющей стадии выражается уравнением
r = k[RX] (2)
В действительности механизм рассматриваемых реакций нуклеофильного замещения является более сложным.
В настоящее время общепринято, что разрыв связи С-Х существенно облегчается или вообще становится возможным лишь за счет взаимодействия замещаемой группы Х с электрофилами. Образование донорно-акцепторной связи I или водородной связи II
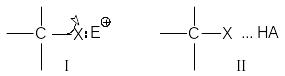
ведет к предварительной поляризации связи С-Х и облегчает ее последующий разрыв. Замещение некоторых групп (-OH, -OR, -SH, -SR, -NH2, -NHR, - NR2) вообще невозможно без их сильного взаимодействия с электрофилами. Например, замещение гидроксильной группы спирта при действии только бромид-аниона невозможно, но эту же реакцию легко осуществить в кислой среде. Предварительное протонирование гидроксильной группы меняет ее характер: в качестве уходящей группы выступает стабильная молекула воды, что делает энергетически выгодным гетеролиз связи С-ОН и облегчает атаку аниона брома
ROH + HBr RO+H2 + Br– [Br- ... R ... O-H2] Rbr + H2O
Аналогичным образом осуществляется превращение простых эфиров, меркантанов и аминов.
Кроме сильных протонных кислот (НСl, H2SO4) катализаторами реакций нуклеофильного замещения являются кислота Льюиса (Ag+, Hg2+, HgCl, HgCl2, SnCl4, AlCl3, SbCl5, Cu2Cl2, BF3 и др.). Их действие зависит от природы отщепляемой группы. Например, протонные кислоты существенно ускоряют замещение О-, S- и N-содержащих групп и мало влияют на скорость замещения галогенов, поскольку последние являются слабыми основаниями и плохо протонируются. Атомы галогенов дают, однако, прочные донорно-акцепторные комплексы с некоторыми кислотами Льюиса, и их образование облегчает разрыв связи С-Hal при последующей реакции замещения. Так, аллилхлорид медленно гидролизуется водой, но в присутствии хлорида меди (I) легко дает аллиловый спирт
![]()
C H2=CH–CH2
Cl
Cu2Cl2
+ H2O
CH2=CH–CH2O+H2
+ Cu2Cl3
H2=CH–CH2
Cl
Cu2Cl2
+ H2O
CH2=CH–CH2O+H2
+ Cu2Cl3
CH2=CH-CH2OH + HCl + Cu2Cl2
При этом может произойти изменение механизма SN2 в некаталитической реакции на SN1 - в каталитической:
медл.
RCl + Cu2Cl2 R+ + Cu2Cl3
R+ + Y- RY
Cu2Cl3– Cu2Cl2 + Cl–
Ослабление связи С-Х и уменьшение энергии переходного состояния в реакциях нуклеофильного замещения происходит и при образовании водородных связей между уходящей группой и протонодонорным агентом.
Так, скорость реакции метанолиза трифенилхлорметана
(C6H5)3CCl + CH3OH (C6H5)3COCH3 + HCl
описывается кинетическим уравнение третьего порядка:
r = k[(C6H5)CCl][CH3OH]2
Это согласуется с механизмом, согласно которому замещение хлора происходит только при условии его специфической сольватации другой молекулой спирта:
(C6H5)3C-Cl + CH3OH (C6H5)3C–ClHOCH3
Образующийся ассоциат дает далее продукт замещения под действием второй молекулы нуклеофила - спирта
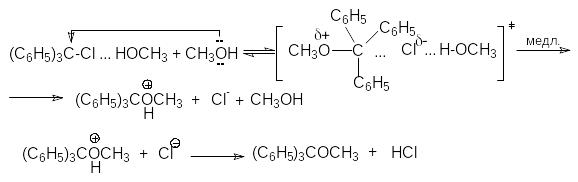
Противоположный эффект наблюдается при специфической сольватации нуклеофила, поскольку в результате образования водородных связей основность последнего утрачивается, что понижает его нуклеофильное сродство к положительно заряженному реакционному центру. При этом отрицательно заряженные нуклеофилы сольватируются сильнее нейтральных молекул или групп. Так, например, в реакции
CH3J + Br– CH3Br + J–
преобладающее влияние имеет сольватация бромид-атома, а не атома иода в молекуле СH3J. Это связано с большей электроотрицательностью брома по сравнению с иодом, что обусловливает большую прочность водородной связи с Br– по сравнению с отщепляющимся J–. В результате скорость этой реакции в метаноле в десятки тысяч раз меньше, чем в апротонных растворителях, где эффект специфической сольватации отсутствует.
Сольватация существенно влияет не только на общую скорость реакции, но и на соотношение механизмов SN1 и SN2. При прочих равных условиях первый из них тем более вероятен, чем выше полярность растворителя и его способность к специфической сольватации уходящей группы и чем выше кислотность катализатора. Такая роль катализаторов связана с выделением энергии сольватации, компенсирующей энергетические затраты на гетеролиз С-Х-связи.
В действительности SN1 и SN2-реакции не отделены друг от друга резкой гранью, между ними имеется область так называемых пограничных механизмов. При “чистом” SN2-механизме с отрицательно заряженным нуклеофилом реакционный центр в переходном состоянии имеет частичный отрицательный заряд, поскольку образование новой связи С-Y опережает разрыв старой связи С-Х. Если на эту реакцию начинают действовать факторы, благоприятствующие SN1-механизму (температура, сильносольватирующая среда, катализаторы –электрофилы) разрыв старой связи начинает опережать образование новой; реакционный центр в переходном состоянии приобретает положительный заряд и в пределе возникает ситуация, когда старая связь практически разорвана, а новая не образовалась, что соответствует чистому механизму SN1. Переход от механизма SN2 к механизму SN1 иллюстрируется следующей схемой: