

Лекции / Kurs_lektsiy_po_patofiziologii_Ch_1_2018_1
.pdfвсех разновидностей апоптоза. Первая стадия отличается большим разнообразием и существенно зависит от типа клеток и сигналов (индукторных факторов).
Показано, что в одних случаях генетическая программа гибели клетки включается внешними факторами, а в других случаях эта программа реализуется при отсутствии соответствующих внешних факторов (табл. 6).
Таблица 6 Разновидности сигналов, приводящих к индукции апоптоза
Происхожде- |
Природа |
Примеры |
|
ние сигнала |
сигнала |
|
|
Внеклеточные |
Антиген |
Негативная селекция ти- |
|
сигналы |
Гормон |
моцитов |
|
|
Цитокин (FasL) |
Действие глюкокортико- |
|
|
|
идов |
|
|
|
Fas-зависимый апоптоз |
|
|
|
Цитолиз, |
вызванный |
|
|
ФНОα |
|
Внутриклеточ- |
Повреждение хро- |
Радиационная гибель |
|
ные сигналы |
мосом. Другие из- |
лимфоцитов в интерфазе |
|
|
менения хроматина |
Действие топоизомераз |
|
Дефицит |
Дефицит ростовых |
Гибель кроветворных |
|
факторов |
факторов |
клеток |
|
|
Дефицит антигенов |
Гибель активированных |
|
|
Дефицит корецеп- |
Т-клеток в отсутствии |
|
|
тора |
ИЛ-2 |
|
|
|
Гибель В-клеток в заро- |
|
|
|
дышевых центрах |
|
|
|
Апоптоз при активации |
|
|
|
Т-клеток в отсутствии |
|
|
|
сигнала с рецептора |
|
|
|
CD28 |
|
Наиболее полно изучены условия экзогенной индукции (внешними воздействиями) апоптоза Т-лимфоцитов, особенно кортикальных тимоцитов. Классические индукторы апоптоза тимоцитов – глюкокортикоиды. Последние после проникновения в клетку проявляют
239
свое действие через рецепторы, локализующиеся в ядре. Одни тимоциты гибнут под действием глюкокортикоидов, а другие – нет (являются гормонорезистентными). Сигнал, поступающий в Т-лимфоциты через рецептор для антигена, в норме может либо приводить к активизации пролиферации клеток, либо вызывать их апоптоз (обозначаемый как активационный).
В качестве сигналов апоптоза могут служить: 1) отсутствие ко-
стимуляции через мембранные молекулы CD28, CD40 и др.; 2) отсутствие ростовых факторов, прежде всего ИЛ-2; 3) предварительное перекрестное сшивание молекул CD4; 4) мембранные молекулы лимфоцитов CD2; 5) молекулы главного комплекса гистосовместимости класса I; 6) β1- и β2-интегрины; 7) ФНОα и т.д.
Сигнал к развитию апоптоза подается через рецептор цитокина ФНОα, который имеет цитоплазматический домен гибели, передающий летальный сигнал внутрь клетки. Некоторые цитокины, например, ИЛ-2 и ИФ-γ, в зависимости от ситуации могут либо индуцировать, либо предотвращать развитие апоптоза. Такая амбивалентность эффектов свойственна и ряду других факторов: ингибиторам синтеза белка, активаторам и ингибиторам активности протеинкиназ и т.д.
Существуют также рецепторы, для которых передача сигнала к развитию апоптоза является основной их функцией. Это Fas-рецептор (АРО-1, CD95) и белки группы DR (Death receptors – «рецепторы смерти»): DR3 (АРО-3), DR4 (TRAIL-R1), DR5 (TRAIL-R2).
Впроцессе передачи внутриклеточного сигнала, приводящего к развитию апоптоза, существуют две фазы (рис. 16).
Первая, более ранняя, фаза может быть различной (она весьма вариабельна), ее характер зависит от вида сигнала – пусковых меха-
низмов (FasL, Fas, TNF, TNFR1 и т.д.).
Следующая, вторая, фаза универсальна для всех разновидностей апоптоза.
Вслучае Fas-зависимого апоптоза связывание Fas-лиганда (FasL)
стримерным Fas-рецептором приводит к конформационным изменениям в цитоплазматическом домене смерти рецептора Fas. Это создает возможность его связывания с аналогичным доменом адапторной молекулы FADD (Fas-associated death domain), a затем – с таким же доменом белка RIP (receptor interacting protein).
240
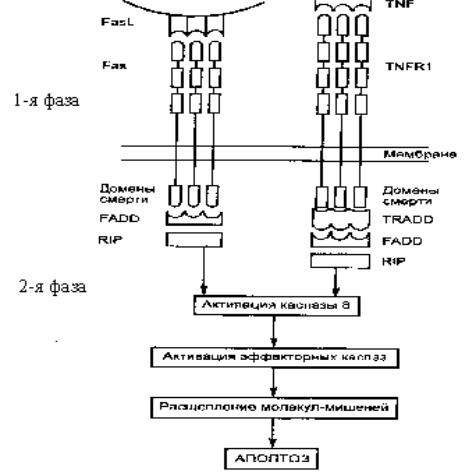
Аналогичные события происходят при действии TNF (ФНОα) через рецептор TNFR1.
Только в этом случае с рецептором взаимодействует адапторный белок TRADD (TNFR-associated death domain), который передает сиг-
нал апоптоза через другие белки (FADD и RIP).
Рис. 16. Молекулярные взаимодействия при индукции апоптоза через Fas и TNF-рецепторы: FasL и TNF – индукторы апоптоза; TNFR1 и Fas – рецепторы сигналов апоптоза; TRADD, FADD, RIP – белки, передающие сигнал апоптоза
Образующийся DISC-комплекс (death-inducing signaling complex)
активирует фермент – каспазу 8, которая относится к эффекторным каспазам, вызывающим расщепление молекул-мишеней и развитие апоптоза.
Выделяют и многие другие сигналы, ответственные за развитие апоптоза (протеинкиназы, протеинфосфатазы, онкосупрессоры р53 и р21, транскрипционные факторы c-mys, c-jun, c-fos, nur-77 и т.д.).
241
Выявлена множественность пусковых воздействий и механизмов и единство конечных механизмов реализации апоптоза. В частности, доказано, что апоптоз может быть индуцирован многими внутриклеточными регуляторными факторами (LCK, ZAP-70, Ca2+, кальциневрин, NFAT, TRADD, FADD, RIP и др.), изменяющими активность каспаз цитоплазмы. Последние через влияние на свои ядерные мишени активируют эндонуклеазы, приводящие к деградации ДНК, и, таким образом, ответственны за универсальный этап индукции апоптоза.
2.2.7. Роль макрофагов в распознавании и удалении апоптотирующих клеток
Различные апоптотирующие клетки, в том числе апоптотирующие гранулоциты и лимфоциты, быстро распознаются и поглощаются макрофагами. Выделяют три основных механизма распознавания и удаления клеток, подвергающихся апоптозу.
Во-первых, макрофаги с помощью лектинов адгезируют различные клетки, вступающие на путь апоптоза. Это происходит в результате специфической перестройки углеводных компонентов мембран апоптотирующих клеток с потерей терминальных сиаловых кислот мембранными гликопротеидами, что приводит к экспрессии сахаров и снижению общего отрицательного потенциала наружной поверхности мембраны.
Во-вторых, распознавание апоптотирующих клеток осуществляется с помощью макрофагального α4β3-интегринового рецептора, а также синтезируемого и секретируемого в микроокружение макрофагами тромбоспондина, выполняющего роль молекулярной «скрепки» между апоптотирующей клеткой и макрофагом.
В-третьих, в распознавании апоптотирующих клеток участвует макрофагальный фосфатидилсериновый рецептор. Уже на ранних этапах программированной гибели в апоптотирующей клетке происходит инверсия мембранных фосфолипидов. Нейтральные фосфолипиды с наружного слоя перемещаются на внутренний, а отрицательно заряженные фосфолипиды с внутреннего слоя мембраны перемещаются на наружный слой. В результате перемещения фосфатидилсерина наружу он распознается специфическим макрофагальным фосфатидилсериновым рецептором.
242
2.3. Роль апоптоза в развитии патологии
За последнее десятилетие число опубликованных работ по проблеме апоптоза увеличилось более чем в 10 раз. Особенно возросло число исследований, посвященных роли апоптоза в развитии многообразной патологии, встречающейся в процессе жизни современного человека.
В зависимости от вида и степени выраженности патологии клетка погибает, как отмечено выше, либо в результате апоптоза, либо некроза. В конечном итоге выбор организмом вида гибели клеток (апоптоз или некроз) зависит от содержания в этих клетках ряда ФАВ: 1) активных форм кислорода (Н2О2, О2–, О–, ОН–), определяемого соотношением активности оксидантной и антиоксидантной (супероксиддисмутаза – СОД, каталаза, селензависимая глутатионпероксидаза, селен, L-карнозин, токоферол, мелатонин) систем; 2) оксида азота (NO), определяемого в основном активностью индуцибельной, самой мощной нитрооксидсинтетазы (iNOS), а также конститутивных эндотелиальной (eNOS) и нейрональной (nNOS) синтетаз; 3) НАД+ и макроэргов (КРФ, АТФ, АДФ, ГТФ, ГДФ) и их соотношения; 4) провоспалительных (ИЛ-1, ИЛ-3, ИЛ-6, ИЛ-8, ФНОα, ИФ-γ, ГМ-КСФ и др.) и противоспалительных (ИЛ-10, ИЛ-4, ИЛ-22, ИЛ-18 и т.д.) цитокинов и их соотношения; 5) белков острой фазы (СРБ, α-антитрипсина, кислого α1-гликопротеина, α2-макроглобулина и др.) и их соотношения.
Доказано, что значительное снижение в клетках концентрации НАД+ и АТФ, повышение уровня активных форм кислорода (особенно Н2О2, О2–), NO и уменьшение количества ферментных и неферментных антиоксидантов, а также существенное повышение содержания провоспалительных цитокинов и белков острой фазы приводит к развитию и усилению некроза и одновременно к угнетению апоптоза.
В последнее время особую роль в развитии как физиологических (в том числе апоптоза), так и патологических (в том числе некроза) процессов отводят важнейшему внутриклеточному медиатору – NO. Он может выполнять роль как молекулы-адаптогена, так и молекулыразрушителя.
Общеизвестно, что в малых (физиологических) количествах NO, определяемых активностью, главным образом, конститутивной нит-
243
рооксидсинтетазы, вызывает физиологические (апоптотические) изменения: активирует гуанилатциклазу, повышает содержание и действие цГМФ, снижает агрегацию тромбоцитов и адгезию нейтрофилов к эндотелию, обеспечивает внутриклеточный кальциевый гомеостаз, а также микробицидный и тумороцидный эффект фагоцитов.
В больших количествах (на два порядка превышающих нормальные значения и достигающих сотен микромолей на 1 кг массы тканей) NO вызывает развитие дизадаптивных и деструктивных изменений, в частности повышение проницаемости стенок сосудов, формирование и увеличение отека в тканях, развитие вазодилатации, кардиотоксическое действие, развитие острой сердечной недостаточности, в итоге приводящие к прогрессирующей, вплоть до необратимой, артериальной гипотензии.
Известно, что резко сниженный уровень NO сопровождается снижением адаптации организма, а резко повышенный его уровень запускает суицидальную программу (самоуничтожение) клеточнотканевых структур. И то, и другое необходимо корригировать, используя для этого различные пути, способы и средства лечения.
Показано, что в условиях различной (и экзогенного, и эндогенного происхождения) патологии неизбежно возникают нарушения тех или иных звеньев, как индукции, так и регуляции процесса апоптоза.
Нарушение апоптоза – обязательный компонент формирования многих патологических процессов. При этом характер и выраженность развития патологического процесса определяется направленностью изменения апоптоза (ослаблением или усилением).
2.3.1. Апоптоз как обязательный компонент развития типовых патологических процессов
Ведущая роль апоптоза в реализации многообразных физиологических процессов, в поддержании оптимального клеточного и тканевого гомеостаза, в обеспечении нормального развития клеточнотканевых структур организма общеизвестна и не вызывает сомнений. Значение же апоптоза в формировании типовых патологических процессов может быть разным: решающим, незначительным и отсутствующим.
В обобщающем варианте роль апоптоза в развитии различной патологии представлена на рис. 17, из которой видно, что апоптоз имеет
244
решающее значение в механизмах стресса, особенно в специфических стрессорных реакциях организма на действие различных интенсивных раздражителей.
|
|
|
|
|
|
|
|
|
Апоптоз |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Участие апоптоза в формиро- |
|
|
|
Изменение выраженности |
|
|
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
вании типовых патологических |
|
|
|
|
|
|
апоптоза |
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
процессов |
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Гибель лимфоци- |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
тов и энтероцитов |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
при стрессе |
|
|
|
|
|
|
Ослабление |
|
|
Усиление |
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Врожденные |
|
|
|
|
|
|
|
|
|
|
|
Повышение ве- |
|
|
|
|
||||
|
|
Гибель клеток |
|
|
|
|
|
|
|
|
дефекты тканей, |
|
|
||||
|
|
|
|
|
|
|
роятности разви- |
|
|
|
|
|
|||||
|
|
при септическом |
|
|
|
|
|
|
|
уродства |
|
|
|||||
|
|
|
|
|
|
|
|
тия злокаче- |
|
|
|
|
|
||||
|
|
шоке |
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
ственных опухо- |
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
Панцитопении, |
|
|
||||
|
|
|
|
|
|
|
|
|
лей |
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
Локальная гибель |
|
|
|
|
|
|
|
|
|
|
|
|
первичные им- |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
мунодефициты |
|
|
|
|
|
клеток при реок- |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
сигенации после |
|
|
|
|
|
|
Сочетание вол- |
|
|
|
|
|
|||
|
|
ишемии |
|
|
|
|
|
|
чаночного и |
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
лимфопро- |
|
|
|
Нейродегенера- |
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
лиферативного |
|
|
|
тивные процес- |
|
|
||
|
|
Повреждение кле- |
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
синдромов |
|
|
|
сы |
|
|
|||
|
|
ток Т-киллерами |
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
при аутоиммун- |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
ных процессах |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Бактериальные ин- |
|
Вирусные ин- |
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
фекции (эффект |
|
фекции, в том |
|
|
||||
|
|
|
|
|
|
|
|
|
суперантигенов) |
|
числе СПИД |
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Замедление гибе- |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
ли клеток- |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
эффекторов в |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
позднюю фазу |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Действие неблагоприятных факторов среды, |
|
|
|||||||
|
|
немедленной ал- |
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
цитотоксической терапии |
|
|
|||||||
|
|
лергии |
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Рис. 17. Роль апоптоза в патологии
Типичное проявление системного стресса – лимфоцитопения и опустошение лимфоидных тканей и органов (тимуса, миндалин, лимфатических узлов, пейеровых бляшек кишечника и др.), обусловленных действием избыточно образующихся при стрессорных реакциях глюкокортикоидов (особенно кортизола). Происходит гибель не только лимфоцитов, но и энтероцитов.
245
Системная апоптотическая гибель клеток наблюдается также при септическом шоке. Важное значение в механизмах данного системного апоптоза имеют гиперпродукция макрофагами и другими клетками ФНОα (TNFα) и активация рецепторов TNFR1. При различных инфекционных процессах апоптотическая гибель клеток зависит в основном от выделяемых микроорганизмами экзотоксинов. Последние в виде суперантигенов выступают в качестве индукторов активационного апоптоза. Сам же инфекционный типовой патологический процесс не является обязательным в развитии апоптоза.
Апоптотическое повреждение клеток, возникающее под действием Т-киллеров, происходит при различных аутоиммунных процессах. Локальную апоптотическую гибель клеток обнаруживают также при постишемической реоксигенации. На начальных этапах развития лихорадки и воспаления апоптоз не является обязательным компонентом в механизмах их развития. Ибо в начале воспалительного процесса гибель клеточно-тканевых структур в очаге воспаления развивается благодаря не апоптозу, а некрозу, сопровождающемуся выходом внутриклеточных структур (в том числе активных гидролаз) наружу, что приводит к последующей некротической гибели соседних клеточных и внеклеточных образований, вплоть до их гнойного расплавления. По мере протекания воспаления, особенно на поздних этапах его развития, процесс апоптоза начинает занимать все большее и большее место в гибели выполнивших свои защитные функции иммунных клеток (в том числе фагоцитов) с целью более быстрого их обновления.
При аллергическом воспалении, особенно в позднюю фазу ГНТ, происходит замедление элиминации, а значит, затруднение гибели эффекторных клеток. Это обусловлено способностью данных клеток к самоподдержанию благодаря выработке аутокринных цитокинов (например, ГМ-КСФ), защищающих эти клетки от апоптоза.
В развитии опухолей и реализации их отрицательного действия на целостный организм участие апоптоза не является обязательным. В то же время доказано, что при угнетении апоптоза значительно замедляется элиминация как генетически измененных собственных клеток, так и поступивших в организм чужеродных клеток. Таким образом, апоптоз может, как быть (что реже), так и не быть (что чаще) обязательным компонентом различных видов патологий (патологических процессов и заболеваний).
246
2.3.2. Патология, связанная с изменением выраженности апоптоза
В настоящее время доказано, что в развитии различных видов патологии могут иметь значение как ослабление процесса апоптоза, так и его усиление.
Патология, обусловленная ослаблением апоптоза
Исходя из важной роли апоптоза в реализации многообразных физиологических процессов, можно считать доказанным, что ослабление апоптоза обязательно, хотя и в различной степени, отражается на процессах роста и дифференцировки клеток, удаления из организма генетически поврежденных клеточных и внеклеточных структур, а также на формировании аутотолерантности. Показано, что с ослаблением апоптоза связано развитие следующих основных групп процессов и заболеваний: 1) различных дефектов развития тканей, органов, систем и частей тела; 2) аутоиммунных процессов и заболеваний; 3) злокачественных опухолей.
Известно большое количество ингибиторов апоптоза как экзогенного, так и эндогенного происхождения. К ним, в частности, относятся липополисахариды грамотрицательных бактерий, вирусные белки (аденовирусы, бакуловирусы, вирус Эпштейна–Барр, герпесвирус и др.), эстрогены, андрогены, нейтральные аминокислоты, ингибиторы протеаз, производные амантадина, рилузол, флупертин, цинк, стимуляторы миелопоэза (Г-КСФ, ГМ-КСФ), интерлейкины (ИЛ-1β, ИЛ-2, ИЛ-3, ИЛ-4, ИЛ-6, ИЛ-8, ИЛ-9, ИЛ-10), интерфероны (главным образом, ИФН-γ), лейкотриены (главным образом, ЛТ-В4).
Важно отметить, что один и тот же цитокин, гормон и регуляторное вещество может быть и ингибитором, и индуктором апоптоза. Это определяется типом клеток-мишеней, степенью их дифференцированности, а также особенностями функционирования внутриклеточных механизмов. Так, например, ИЛ-4 и ИЛ-10 ингибируют апоптоз Т- и В-лимфоцитов и индуцируют апоптоз циркулирующих моноцитов. Тестостерон для клеток предстательной железы является ингибитором апоптоза, для фолликулярных клеток яичника – индуктором. Эстрадиол в начале менструального цикла ингибирует апоптоз клеток эндометрия матки, в конце цикла – индуцирует апоптоз этих клеток.
247
Ослабление апоптоза при аутоиммунных заболеваниях. Экспе-
риментальные исследования показали, что ослабление апоптоза путем трансфекции мышей геном bcl-2, приводящей к гиперпродукции в различных клетках организма фактора Bcl-2, сопровождается развитием системных аутоиммунных процессов, вплоть до системного красноволчаночного синдрома. Одновременная гиперэкспрессия гена c-myc у трансфектированных мышей, кроме аутоиммунных процессов, приводила также к лимфопролиферативным процессам.
Мутации гена рецептора Fas и гена Fas-лиганда у мышей линии MRL также сопровождаются развитием системных аутоиммунных процессов, в частности системной красной волчанки, а также гломерулонефрита, незлокачественного лимфо-пролиферативного синдрома (проявляющегося гипертрофией периферических лимфоидных образований с накоплением в них Т-лимфоцитов). Эти же нарушения возникают при повышенной продукции фактора Bcl-2, ограничивающего апоптоз. От экспрессии генов fas и bcl-2 в большей степени зависит селекция клонов Т-лимфоцитов в периферической крови, чем в тимусе.
При ревматоидном артрите Т-лимфоциты в суставной полости не подвергаются апоптозу (несмотря на высокую продукцию рецептора Fas и низкую – фактора Bcl-2), тогда как нейтрофилы в тех же пораженных воспалительным процессом суставных полостях подвергаются массированному апоптозу. При ревматоидном артрите усиливается активационный апоптоз Т-лимфоцитов в периферической крови.
Обнаружено, что образующиеся при системной красной волчанке аутоантитела одновременно являются мишенями действия каспаз, обуславливающих включение эффекторного механизма апоптоза, и угнетают активированные клеточным стрессом серин-треониновые киназы.
Выявлена важная роль ослабления апоптоза в патогенезе следующих основных аутоиммунных заболеваний щитовидной железы:
гипотиреоз, характеризующийся лимфоидной инфильтрацией железы и образованием комплементопосредованных аутоантител к таким тиреоидным антигенам как рецептор ТТГ, тиреоглобулин, тиреоидная пероксидаза;
гипертиреоз в виде болезни Грейвса – диффузного токсического зоба, который проявляется активацией рецептора ТТГ (являющегося специфическим аутоантигеном) под действием образующихся ауто-
248