

Лекции / Kurs_lektsiy_po_patofiziologii_Ch_1_2018_1
.pdfантител (в данном случае ослабление апоптоза обусловливает увеличение количества тиреоцитов в железе);
гипотиреоз в виде тиреоидита Хашимото – хронического лимфоцитарного тиреоидита, который характеризуется деструктивными изменениями и уменьшением функционально активного фолликулярного эпителия щитовидной железы (вследствие высокой активности интратиреоидных лимфоцитов-киллеров. Один из надежных диагностических критериев аутоиммунного тиреоидита (даже до развития клинических проявлений гипотиреоза) – оценка степени снижения экспрессии рецептора Fas на тиреоцитах (взятых из пунктата щитовидной железы) и увеличения в крови количества тиреоидных аутоантител.
Ослабление апоптоза при злокачественных опухолях. Доказано,
что различное по степени и длительности угнетение процесса апоптоза сопровождается развитием различных сόлидных и особенно гематогенных злокачественных опухолей. Особо важная роль в их развитии принадлежит соматическим мутациям или дефициту гена р53. В клетках с нерепарированными разрывами цепей ДНК при участии нормального гена р53 формируется сигнал к развитию апоптоза. В большинстве злокачественно трансформированных клеток образуется мутантная (аномальная) форма гена р53, неспособная индуцировать апоптоз. Такие клетки утрачивают нормальные связи с межклеточным матриксом и другими клетками микроокружения, не подвергаются апоптозу, а продолжают интенсивно делиться и метастазировать.
В основе развития некоторых типов лимфом лежат транслокации гена bcl-2, когда он перемещается из хромосомы 18 к гену IgН хромосомы 14. Следствие этих хромосомных транслокаций – гиперэкспрессия гена bcl-2 и повышение резистентности генетически измененных клеток к индукции апоптоза.
Показано, что под влиянием различных онкогенов (особенно bcl- 2) опухолевые клетки становятся резистентными к действию различных физических и химических (в том числе химиотерапевтических) веществ, которые в нормальных клетках всегда индуцируют апоптоз, т.е. в опухолевых клетках процессы апоптоза обычно подавляются благодаря антиапоптозному действию некоторых генов (bcl-2, c-fes и
др.).
249
Вто же время известно, что гиперэкспрессия онкогенов (например, c-myc) в зависимости от условий приводит к активизации либо пролиферации, либо апоптозной гибели клеток. Последняя обусловлена действием различных физиологически активных компонентов цитотоксических гранул, особенно перфоринов и гранзимов, которые при совместном действии способны активировать продукцию серинэстеразы, ответственной за индукцию фрагментации ДНК клетокмишеней и в целом за индукцию процесса их апоптоза. В гиперплазированной (опухолевой) ткани возможен и прямой, независимый от действия цитотоксических гранул, механизм развития апоптоза клеток и регрессии опухоли через молекулы рецептора Fas, расположенные на поверхности клеток.
Апоптоз в опухолевых клетках может активироваться под действием γ-излучения, УФО, гипертермии, гипотермии, под влиянием цитокинов ИЛ-4, ИЛ-10, ФНОα и др., а также под действием ряда химиотерапевтических препаратов (цисплатин, этопозид, тенипозид и др.). Однако способность опухолевых клеток к апоптозу под влиянием указанных выше средств неустойчива и ограниченна.
Внастоящее время считают доказанным, что и трансформация, и прогрессирующий рост клеточного клона зависят не только от онкогенных сигналов, но и от дополнительных антиапоптозных сигналов, обусловленных активностью внутриклеточных ингибиторов апоптоза, т.е. апоптоз играет определенную контролирующую роль в противоопухолевой защите.
Важную роль в угнетении апоптоза в опухолевых клетках играют (кроме отмеченных выше) следующие патогенетические факторы:
1)снижение содержания НАД+ и АТФ; 2) повышение образования активных форм кислорода (Н2О2, О2 – и др.); 3) увеличение продукции NO (в результате резкой активации iNOS); 4) уменьшение активности антиоксидантных ферментов и количества антиоксидантных витаминов и микроэлементов.
Чем выраженнее эти внутриклеточные сдвиги, тем более прогрессивно развивается опухоль. Одновременно отмечено, что усиленно образующиеся в опухоли Н2О2, О2–, ОН– и NO существенно ослабляют противоопухолевый эффект цитостатиков и способствуют развитию некроза опухолевых клеток.
Н2О2 угнетает апоптоз, препятствуя захвату макрофагами апоптотически измененных клеток. Избыток свободных радикалов, угнетая
250
продукцию АТФ в клетке, также сопровождается блокадой различных звеньев апоптоза (выхода на поверхность клетки молекул фосфатидилсерина, активации каспаз).
Антиоксиданты не только уменьшают количество свободных радикалов, но и способны даже в присутствии Н2О2 поддерживать необходимый для жизнедеятельности клеток уровень АТФ. Более того, на фоне введенных в организм антиоксидантов усиливается противоопухолевый эффект цитостатиков в основном за счет активизации процесса апоптоза. Показано, что в отсутствие Н2О2 цитостатики проявляют максимальный противоопухолевый эффект, усиливая гибель опухолевых клеток путем активации апоптоза.
Избыточно образующийся при опухолевом (оксидативном) стрессе NO, приводит у онкологических больных к выраженным как цитотоксическим, так и гипотензивным эффектам. Эти нежелательные клинические эффекты NO можно существенно ослабить путем применения следующих препаратов: 1) цианкобаламина в больших дозах (10 мг/кг); 2) глюкокортикоидов, способных ингибировать iNOS; 3) аргиназы, способной разрушать предшественник NO – L- аргинин.
В заключение следует отметить, что нарушение или блокада механизмов апоптоза способствуют развитию и ускорению роста злокачественных опухолей.
Патология, обусловленная усилением апоптоза
Важное место в патологии занимают заболевания, основу которых составляет усиление процесса апоптоза самых разных клеток организма (табл. 7).
Обнаружено большое количество индукторов апоптоза как экзогенного, так и эндогенного происхождения. В частности, к ним относятся стрессогенные факторы, ионизирующие излучения (УФ-лучи, γ- лучи), химиотерапевтические средства (цисплатин, доксорубицин, метотрексат, винкристин и др.), ингибиторы белкового синтеза (например, циклогексимид), этанол, оксиданты (Н2О2, свободные радикалы и др.), сульфиды, гипертермия, белки теплового шока, протеазы, β-амилоидный пептид (предшественник амилоидного белка), глюкокортикоидные гормоны, ионы кальция, глутамат, аспартат, дофамин.
251

Таблица 7 Заболевания, связанные с усилением апоптоза
|
Группа |
Конкретные |
Комментарии |
|
|
заболеваний |
заболевания |
|
|
|
|
|
|
|
|
Врождённые ано- |
Синдром Дауна и др. |
Преобладание апоптоза |
|
|
малии |
|
при формировании ло- |
|
|
|
|
кальных структур |
|
|
|
|
|
|
|
Болезни крови |
Миелодисплазии, апла- |
Усиление апоптоза кле- |
|
|
(цитопении) |
стическая, железо, фо- |
ток отдельных или всех |
|
|
|
лат-, В12- дефицитные |
ростков кроветворения в |
|
|
|
анемии, тромбоцитопе- |
процессе их развития |
|
|
|
ния, нейтропения |
|
|
|
|
|
|
|
|
Инфекционные |
Различные инфекцион- |
Апоптоз клеток иммун- |
|
|
(бактериальные) |
ные процессы, сепсис |
ной системы вызывают |
|
|
заболевания |
|
суперантигены, токси- |
|
|
|
|
ны; при сепсисе – |
|
|
|
|
накапливающийся в |
|
|
|
|
крови ФНОα |
|
|
|
|
|
|
|
Вирусные |
Различные вирусные за- |
Индукторами апоптоза |
|
|
инфекции |
болевания (СПИД и др.) |
при СПИДе служат ви- |
|
|
|
|
русные факторы, в част- |
|
|
|
|
ности gpl20, взаимодей- |
|
|
|
|
ствующий с CD4 |
|
|
|
|
|
|
|
Дегенеративные и |
Боковой амиотрофиче- |
Усиление апоптоза |
|
|
атрофические за- |
ский склероз, болезнь |
нейронов и других кле- |
|
|
болевания нерв- |
Альцгеймера, спиналь- |
ток в определённых |
|
|
ной системы |
ная мышечная атрофия |
участках центральной |
|
|
|
|
нервной системы |
|
|
|
|
|
|
|
Другие |
Инфаркт миокарда |
Преобладание апоптоза |
|
|
заболевания |
|
кардиомиоцитов в пери- |
|
|
|
|
од «реперфузии» мио- |
|
|
|
|
карда |
|
|
|
|
|
|
|
|
Токсические гепатиты |
Апоптоз гепатоцитов |
|
|
|
|
под действием различ- |
|
|
|
|
ных ядов, в том числе |
|
|
|
|
этанола |
|
|
|
|
|
|
|
|
|
|
|
|
|
252 |
|
|
Некоторые интерлейкины также способны индуцировать апоптоз соответствующих клеток. Так, ИЛ-1 индуцирует апоптоз клеток поджелудочной железы, ИЛ-10 – периферических моноцитов, ИЛ-12 – натуральных киллеров и т.д. В конце менструального цикла и эстрогены, и прогестерон индуцируют апоптоз клеток эндометрия матки.
Гибель клеток путем апоптоза чаще обнаруживается:
в гормонально-зависимых тканях (например, гибель клетокмишеней после удаления соответствующих эндокринных желез, в частности гибель клеток предстательной железы после кастрации или гибель клеток коры надпочечников после угнетения продукции АКТГ гипофизом и т.д.);
при снижении кровоснабжения (развитии ишемии) органа;
при появлении дефектных как пролиферирующих (эпителий кишечника), так и непролиферирующих (иммунная система) клеток, возникающих в результате мутаций, вызванных действием вирусов, радиации и т.д.
Кзаболеваниям, характеризующимся усилением апоптоза, отно-
сятся: 1) врожденные соматические аномалии (уродства, дефекты структур, основу которых составляют изъязвления, т.е. «минус ткани»); 2) атрофические или апластические, дегенеративные или дистрофические заболевания нервной, кроветворной, пищеварительной и других систем; 3) ишемические и токсические заболевания жизненно важных органов (сердца, печени, почек и др.); 4) инфекционные (вирусные, бактериальные, паразитарные) заболевания; 5) внутриутробная задержка развития или гибели эмбриона; 6) врожденные поражения сердца; 7) развитие различных дегенеративно-атрофических заболеваний ЦНС (болезнь Альцгеймера, болезнь Паркинсона, боковой амиотрофический склероз, спинальная мышечная атрофия, синдром Дауна, хорея Гентингтона, гормонально-зависимые опухоли (органов репродуктивной системы), преждевременное старение ЦНС и др.).
В патогенезе всех дегенеративных заболеваний ЦНС большое значение имеет снижение устойчивости нейронов определенных структур мозга к стимуляторам апоптоза: эксайтоаминокислотам, вирусным белкам, ионам кальция и др.
253
Характеристика дегенеративно-атрофических заболеваний ЦНС, обусловленных активацией апоптоза
Болезнь Альцгеймера характеризуется накоплением в ускоренно стареющих нейронах коры головного мозга (преимущественно те- менно-височно-затылочных областей), пептида Аа, Р-амилоида – предшественника амилоидного белка (amyloid precursos protein, АРР), глутамата и ионов кальция, способствующих развитию апоптоза этих нейронов. В настоящее время идентифицирован ген болезни Альцгеймера, продукт которого оказался транскрипционным фактором АLG-2. Установлено также, что в механизмах ускорения апоптоза принимает участие активация NMDA-рецепторов и каспаз. Прогрессирующая дегенерация клеток коры мозга обусловлена преимущественно повреждающим действием активных метаболитов кислорода и недостаточностью холинергических терминалей этих нейронов.
Болезнь Паркинсона развивается в результате дегенеративных изменений нейронов подкорковых образований головного мозга, особенно стриатума и компактной части черного вещества. Развитие апоптоза нейронов в этих образованиях мозга обусловлено: 1) блокированием l-митохондриального комплекса, 2) снижением содержания АТФ; 3) уменьшением образования важного антиоксиданта
– глутатиона; 4) формированием окислительного стресса, 5) активизацией NMDA-рецепторов; 6) повышением входа кальция в клетки, 6) усиленной экспрессией гена р53.
Установлено, что назначение дофамина и препарата леводопы активирует, а ингибиторов моноаминоксидазы (L-депрепил) – ингибирует апоптоз.
Боковой амиотрофический склероз, характеризующийся актива-
цией апоптоза двигательных нейронов головного и спинного мозга, проявляется: 1) стимуляцией в них оксидантной системы, возникающей в результате мутации гена COD1; 2) возрастанием концентрации эксайтоаминокислот; 3) увеличением содержания в нервной ткани свободных радикалов; 4) увеличением числа постсинаптических рецепторов к ним и содержанием внутриклеточного кальция в мотонейронах.
Синдром Дауна, наряду с умственной отсталостью, проявляется нарушением (извращением) дифференцировки и уменьшением количества нейронов ЦНС. При данном синдроме, как и при болезни Аль-
254
цгеймера, отмечается отложение амилоида внутри нейронов ЦНС. При синдроме Дауна, как и при болезни Паркинсона и боковом амиотрофическом склерозе происходит внутриклеточное увеличение концентрации свободных радикалов и продуктов ПОЛ.
Хорея Гентингтона, проявляющаяся активизацией апоптоза, развивается в результате мутации гена, кодирующего белок хантингтин, играющий важную роль в обеспечении нормальной структуры и функции нейронов.
Распространенными вариантами патологии, развивающейся в сформировавшемся организме в связи с усилением апоптоза, являют-
ся аплазии и дегенеративные процессы.
При патологии системы крови обнаружены наиболее разнообразные ее формы. Чаще всего они развиваются вследствие недостаточности факторов выживания костномозговых клетокпредшественников. Так, в эксперименте направленная инактивация гена ИЛ-7 приводит к тотальной лимфопении, в значительной степени связанной с гибелью предшественников В- и Т-лимфоцитов на стадиях, предшествующих формированию антигенраспознающих рецепторов.
Аналогичная патология, связанная с мутацией гена общей цепи рецепторов для ИЛ-7, ИЛ-2, ИЛ-4, ИЛ-9 и ИЛ-15, описана у человека как тяжелый комбинированный иммунодефицит.
Сходные причины лежат в основе апластической анемии, анемии при дефиците железа, фолатов, витамина В12, талассемии, тромбоцитопении, нейтропении и панцитопении. Показано, что в патогенезе миелодиспластических процессов, приводящих к панцитопении, важное значение имеет активация апоптоза стволовых клеток и ранних кроветворных предшественников. Повышенная готовность к развитию апоптоза Т-лимфоцитов обнаружена при мультицентрической болезни Кастлемана.
Инфекционные процессы образуют большую группу заболеваний, связанных с усилением апоптоза. Индукторами апоптоза служат бактериальные эндотоксины (например, липополисахарид кишечных микроорганизмов) и экзотоксины (в частности, стафилококков и т.д.). Последние, как правило, выступают в роли суперантигенов, вызывая массовую пролиферацию Т-лимфоцитов с их последующей гибелью по механизму апоптоза. Массовый апоптоз, опосредованный факто-
255
ром некроза опухоли и его рецепторами 1-го типа, развивается при сепсисе.
При вирусных инфекциях сосуществуют факторы, индуцирующие и ингибирующие апоптоз (вирусам принципиально «невыгодна» тотальная гибель клеток-мишеней). Особая ситуация складывается при СПИДе. Установлено, что доля инфицированных клеток среди гибнущих Т-лимфоцитов невелика. Гибель лимфоцитов происходит по механизму апоптоза. Ее выраженность коррелирует с быстрым прогрессированием заболевания. Апоптозу подвергаются предварительно активированные лимфоциты, в основном несущие маркер клеток памяти CD45RO. Полагают, что одним из механизмов, повышающих их чувствительность к активационному апоптозу, может быть перекрестное связывание молекул CD4 мембранным гликопротеином ВИЧ-1 gpl20 (что моделируется действием на Т-клетки моноклональных антител к CD4), а также связывание gpl20 с рецепторами хемокинов. Вирусный белок Tat способен вызывать апоптоз и сенсибилизировать клетки к его индукторам. Определенную роль в активационном апоптозе клеток CD4+ играет корецептор CD28, поскольку антитела к этой молекуле предотвращают гибель Т-клеток больных СПИДом. Более того, клетки CD4+, инфицированные вирусом ВИЧ-1, менее чувствительны к индукции апоптоза, поскольку один из вирусных белков, Nef, подавляет его развитие.
Существует ряд заболеваний, при которых в реализации основного повреждения клеточных структур органа апоптозу принадлежит решающая роль, в частности:
1) инфаркт миокарда (апоптоз является преобладающей формой гибели кардиомиоцитов в период «реперфузии» участка миокарда, ранее подвергшегося ишемии); 2) токсический (в частности, алко-
гольный) и вирусные гепатиты и т.д.
При хронических гепатитах В и С, наряду с прямым проапопто-
тическим действием вирусов на гепатоциты, гранулоциты и лимфоциты, выявлены иммуноопосредованные (как специфические, так и неспецифические) повреждения инфицированных вирусом клеток
(рис. 18).
В зависимости от характера и степени выраженности апоптоза, как видно из рис.18, может либо наступить выздоровление (в результате разрушения инфицированных клеток с помощью перфорингранзимового комплекса и системы Fas / FasL и элиминации их с по-
256
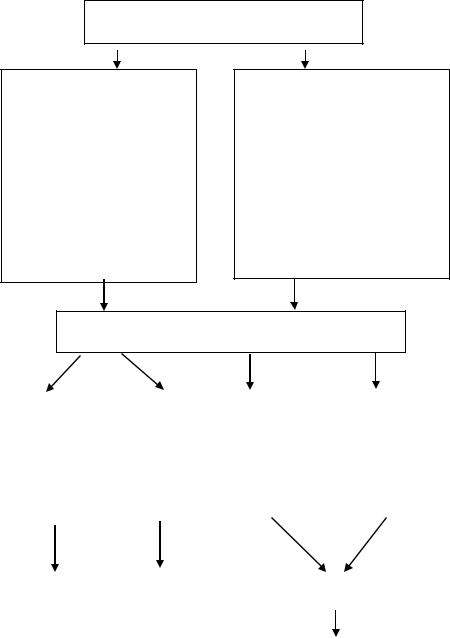
мощью макрофагов), либо развиться хронический гепатит (в результате обширной гибели гепатоцитов) с переходом в цирроз печени и снижение иммунных (как неспецифических, так и специфических) функций организма, приводящее в последующем к активизации патогенного действия вирусов и развитию вторичной инфекции.
Механизмы индукции апоптоза клеток при хронических вирусных гепатитах
Прямое повреждающее действие вирусов гепатита В и С:
–прямое проапоптотическое действие белков рХ (HBV)
и core-белка (HCV)
–повышение чувствительности клеток к проаптотическим стимулам
–активация клеточных факторов транскрипции
–повышение экспресии Fas
Иммуноопосредованное повреждение инфицированных вирусом клеток
Неспецифическое звено иммунитета:
система интерферона
макрофаги, нейтрофилы
АФК, медиаторы воспаления.
Вирус-специфический иммунный ответ:
Система перфорин/гранзим
Система Fas/FasL;
TNF
АПОПТОЗ
|
|
|
|
|
|
ослабление неспецифического иммунного ответа |
|
ослабление вирусспецифического иммунного ответа |
элиминация инфицированных вирусом клеток |
|
обширная гибель |
клеток паренхимы |
печени |
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Выздоровление |
Хронический |
Снижение иммунологиче- |
|
гепатит, |
ских функций организма |
||
|
|||
|
циррроз |
|
|
|
|
Персистенция вирусов, хро- |
|
|
|
низация инфекции |
Рис. 18. Роль апоптоза в патогенезе хронических вирусных гепатитов В и С
При воспалительных заболеваниях на фоне активной миграции лейкоцитов в ткани и повреждения клеточно-тканевых структур за-
257
медляется апоптоз гранулоцитов и мононуклеаров. А при активизации апоптоза иммунокомпетентных клеток и клеток органовмишеней процесс воспаления замедляется и разрешается. В частности, активизации апоптоза Т- и В-лимфоцитов, эозинофилов и нейтрофилов при воспалении бронхов и одновременно повышения жизнеспособности бронхиальных эпителиоцитов можно добиться путем введения глюкокортикоидных гормонов. Противовоспалительный эффект теофиллина также реализуется через стимуляцию апоптоза эозинофилов в ткани легких не только через увеличение образования цАМФ в ткани, но и через супрессию белков семейства Вcl-2.
Роль апоптоза в изменении деятельности гормональнозависимых тканей репродуктивной системы (яичников и эндомет-
рия). Апоптоз в яичниках способствует редуцированию количества ооцитов в процессе жизни женского организма. Так, если яичник новорожденной содержит около 5 млн. ооцитов, то к началу репродуктивного периода их сохраняется только около 300 тыс. Для осуществления менструальной и репродуктивной функций необходимо не более 400 примордиальных фолликулов. Благодаря апоптозу основная масса фолликулов редуцируется путем атрезии фолликулов, происходящей на всех стадиях их развития. Только очень незначительная часть фолликулов проходит полный цикл развития от примордиального до преовуляторного и участвует в процессе овуляции. Результатом усиленного апоптоза в фолликулах на одной или нескольких стадиях их развития может явиться развитие преждевременной недостаточности яичников (синдрома истощения яичников).
Образующееся после овуляции желтое тело при отсутствии беременности также подвергается регрессии благодаря апоптозу.
Влияние внешних апоптогенных факторов на развитие различ-
ных патологических процессов. Первое место среди факторов, усиливающих апоптоз, занимает ионизирующая радиация. В связи с тем, что она индуцирует апоптоз преимущественно лимфоидных клеток, эта сторона ее действия проявляется в иммунной недостаточности, хотя вызываемые облучением нарушения кроветворения, по крайней мере, частично, обусловлены индукцией апоптоза клетокпредшественников. Аналогичным эффектом обладают многие хи-
миотерапевтические препараты (цисплатин, доксорубицин, мето-
трексат, винкристин и др.), используемые при лечении опухолей, а
258