

2.4. Изучение фотохимических превращений региоизомерных диазокетонов 1а, 2а
Диазокетоны 1а,б и 2а,б имеют в УФ спектрах несколько интенсивных полос поглощения в районе 230-350нм, а также более слабую полосу в области 400 нм (табл. 2). На основании обычных критериев отнесения полос поглощения определенным электронным переходам (интенсивность полосы поглощения, смещение в “синюю” или “красную” области спектра при переходе от неполярного к полярному растворителю [48]) можно с определенной долей уверенности сделать отнесение n-π* переходам только длинноволновых полос поглощения диазокетонов 1а,б и 2а,б, наблюдаемых в области 391-414 нм. У этих слабых полос при переходе от неполярного гексана к полярным растворителям (ТГФ, МеОН. СН3СN) наблюдается значительный синий сдвиг полосы поглощения в коротковолновую область спектра (на 10-22 нм, табл. 2).
Таблица 2. Характеристики УФ спектров диазокетонов 1а,б и 2а,б
Диазокетон №№ |
Гексан |
ТГФ |
Метанол |
Ацетонитрил |
||||
1а |
λмакс, нм |
lgε |
λмакс, нм |
lgε |
λмакс, нм |
lgε |
λмакс, нм |
lgε |
251 |
4.21 |
253 |
4.28 |
252 |
4.17 |
252 |
4.11 |
|
303 |
2.85 |
298 |
2.93 |
298 |
3.25 |
298 |
3.1 |
|
413 |
1.46 |
401 |
1.49 |
400 |
1.54 |
400 |
1.46 |
|
2а |
256 |
4.45 |
255 |
4.26 |
254 |
4.26 |
253 |
4.07 |
307 |
3.22 |
304 |
3.37 |
302 |
3.5 |
302 |
3.27 |
|
413 |
1.43 |
392 |
1.48 |
392 |
1.56 |
391 |
1.28 |
|
1б |
235 |
4.22 |
|
|
|
|
|
|
254 |
4.17 |
249 |
4.2 |
231 |
4.34 |
230 |
4.38 |
|
278 |
3.83 |
282*) |
3.78 |
247 |
4.18 |
251 |
4.08 |
|
303 |
3.35 |
305*) |
3.26 |
298*) |
3.43 |
303*) |
3.13 |
|
416 |
1.53 |
414 |
1.56 |
401 |
1.57 |
404 |
1.46 |
|
2б |
230 |
3.57 |
|
|
|
|
231 |
4.37 |
260 |
3.52 |
|
|
|
|
256 |
4.32 |
|
307*) |
2.41 |
|
|
|
|
307*) |
3.42 |
|
410 |
0.4 |
|
|
|
|
400 |
1.32 |
|
*) Перегиб на кривой поглощения
Что же касается полос в области 230-350 нм, которые, очевидно, являются суперпозицией полос поглощения диазокарбонильного фрагмента молекулы [49] и ароматических ядер [48], то сделать их отнесение определенным электронным переходам на основании имеющихся данных в настоящее время не представляется возможным.
Фотолиз диазокетонов изучали на примере пары региоизомерных диазосоединений 1а, 2а (R = F). Для выяснения основных направлений фотолиза, на данном этапе исследования УФ облучение проводили полным светом ртутной лампы среднего давления «Hanau S-81» (100-130 вт) через кварцевый фильтр (λ > 210 нм, т.н. “коротковолновый” фотолиз) без какой-либо дополнительной монохроматизации излучения лампы. Фотохимическую реакцию проводили при температуре 15-20 оС в растворе тетрагидрофурана с добавлением 20-ти эквивалентов МеОН (по отношению к диазокетону) до исчезновения диазокетонов 1а, 2а в реакционной смеси (по данным ТСХ). Продолжительность облучения в этих условиях обычно составляла 3 часа. По окончании фотолиза растворитель и другие легколетучие компоненты отгоняли полностью в вакууме, остаток разделяли на колонке с силикагелем и таким образом определяли препаративные выходы полученных соединений (см. схемы 10, 11 и экспериментальную часть).
Для количественной оценки воспроизводимости полученных результатов каждый фотолиз повторяли еще два раза в аналитическом варианте с меньшим количеством исходных диазокетонов (100-120 мг). После обычной обработки реакционной смеси в этих опытах содержание продуктов реакции определяли методом внутреннего стандарта с N-метилмалеимидом. Структуру выделенных веществ устанавливали с помощью спектров ЯМР 1Н и 13С, а также сопоставлением полученных данных с известными характеристиками их аналогов, полученных при изучении фотохимических превращений региоизомерных диазокетонов 1в, 2в (R = Н) [46]. Структура, некоторые спектральные характеристики и выходы полученных соединений в различных условиях фотолиза приведены на схемах 9, 10 и в экспериментальной части. Молекулярная структура ключевого продукта фотолиза – гидразона 9 по данным РСА приведена на рис. 3.
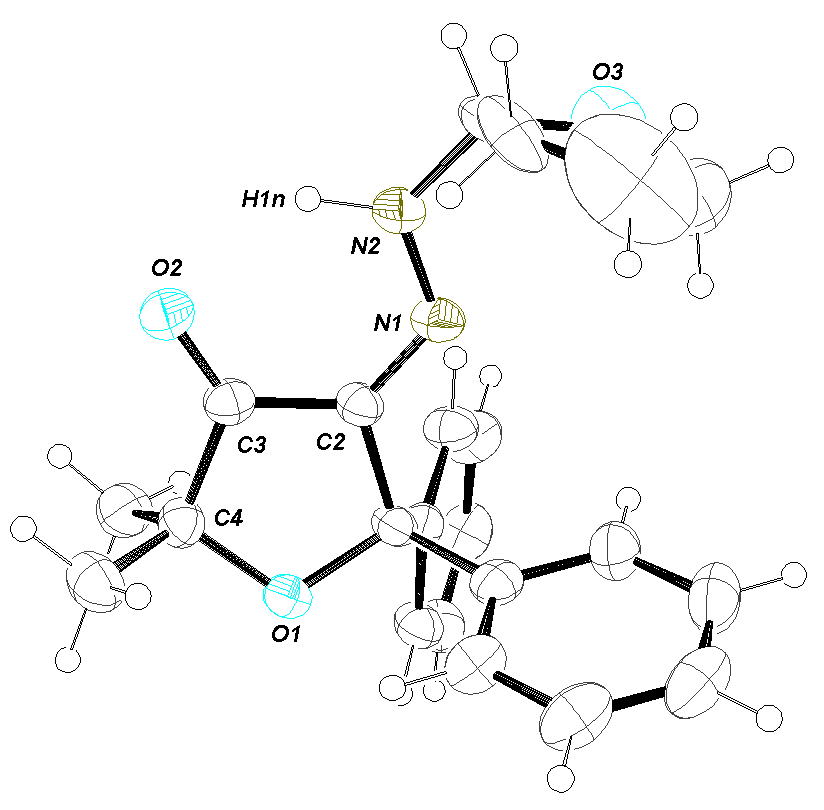
Рис. 3. Молекулярная структура продукта С-Н-внедрения 9в по данным РСА.
Как видно из полученных данных (Схема 9) в результате фотолиза диазокетона 1а в растворе ТГФ/МеОН в течение 3-х часов образуются четыре продукта реакции – 8, 9а, 10а, 4. Основными из них являются метиловый эфир оксетанкарбоновой кислоты 8 (41%) и замещенный гидразон 9а (15%), который формально, как уже отмечалось выше, можно рассматривать как продукт внедрения терминального атома азота диазогруппы в α-СН связь тетрагидрофурана. Наряду с этими соединениями в реакционной смеси были идентифицированы также продукты 1,2-миграции арильной группы 10а и монокетон 4.

Условия фотолиза |
Выходы, % *) |
|||
8 |
9а |
10а |
4 |
|
ТГФ/MeOH, 3 ч |
41 (33) |
15 (14) |
6 |
8 |
ТГФ/MeOH, 1,5 ч |
35 |
28 |
6 |
7 |
ТГФ/MeOH, 3 ч; O2 |
32 |
26 |
8 |
9 |
*) В скобках приведены препаративные выходы продуктов фотолиза
Схема 9. Фотолиз диазокетона 1а
При сокращении времени облучения диазокетона 1а в два раза (с 3-х до полутора часов) выход продукта внедрения 9а увеличивается почти в два раза (с 15 до 28%) при небольшом уменьшении выхода метилового эфира 8 (п. Вольфа) (на 5-6%), тогда как выходы остальных продуктов реакции остаются практически неизменными. По-видимому, гидразон 9а фотохимически нестабилен и в условиях коротковолнового облучения реакционной смеси претерпевает дальнейшие фотохимические превращения.
Фотолиз диазокетона 1а в тех же условиях (ТГФ/МеОН, 3 часа), но в присутствии кислорода приводит к увеличению почти в два раза выхода продукта внедрения 9а при одновременном уменьшении на 9% выхода продукта перегруппировки Вольфа 8. Что касается других продуктов реакции 10а и 4, то их выходы при этом фактически не изменяются (Схема 9).
Результаты фотохимической реакции изомерного диазокетона 2а с двумя геминальными арильными заместителями при α-карбонильной группе оказался практически идентичным фотолизу региоизомера 1а. В результате этого фотохимического превращения тоже образуются четыре продукта реакции, имеющие аналогичную структуру, примерно те же выходы и соотношение, что и при фотолизе диазокетона 1а (Схема 10).
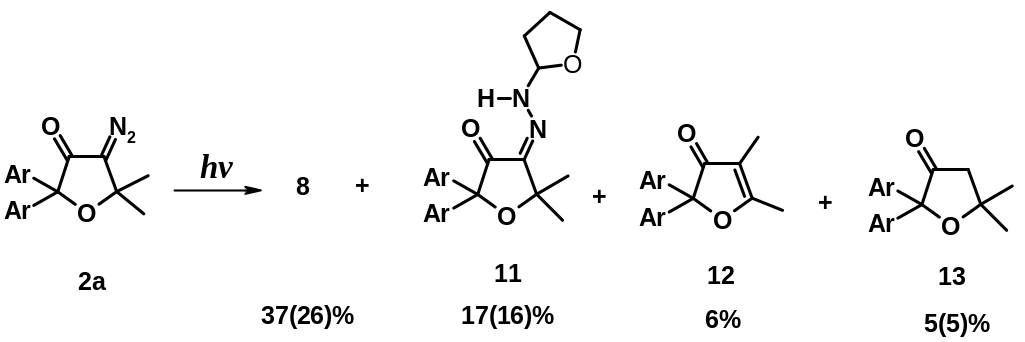
*) В скобках приведены препаративные выходы продуктов фотолиза
Схема 10. Фотолиз диазокетона 2а
Таким образом, можно заключить, что при коротковолновом немонохроматическом облучении региоизомерные диазокетоны 1а, 2а претерпевают практически одни и те же фотохимические превращения, и в этих условиях фотолиза различия в их структуре практически не сказываются на направлении и соотношении наблюдаемых фотопроцессов. Это заключение вполне согласуется с данными расчетов энергии первого возбужденного состояния S1, сделанного на основании характеристик самой длинноволновой полосы поглощения в УФ спектрах региоизомерных диазокетонов 1а и 2а (413-414 нм; табл. 2). Согласно расчетным данным энергия возбужденного состояния S1 у этих диазокетонов примерно одинакова и составляет 69 и 71 ккал/моль соответственно.
2.4. Пути фотохимических превращений региоизомерных диазокетонов 1 и 2
Суммируя полученные нами результаты, а также опираясь на известные литератур-ные данные, касающиеся фотохимических превращений диазосоединений [41,50], можно предложить следующую схему протекания наблюдаемых нами процессов.
При фотохимическом возбуждении диазокетоны 1,2 переходят в синглетное возбужденное состояние 1S1-1,2 (Схема 11), которые частично путем интеркомбинацион-ной конверсии (ИКК) превращается в триплетное, одним из электронных состояний которого является бирадикал с неспаренными электронами на N- и O-атомах диазокарбонильного фрагмента молекулы (CO-CN2). В результате последующего элиминирования азота синглетные диазокетоны 1S1-1,2 дают синглетные ацилкарбены 1S1, которые частично тоже могут переходить в триплетное состояние (3T1) при интеркомбинационной конверсии.

Схема 11.
Диазокетоны 1S1-1,2 в синглетном возбужденном состоянии по согласованному пути или через ацилкарбены 1S1 дают продукты перегруппировки Вольфа и алкильного (арильного) 1,2-сдвига [41,45,47], тогда как триплетные ацилкарбены 3T1, обычно обладающие радикальными свойствами [41,50], отрывают два водородных атома от растворителя, превращаясь в монокетоны 4 или 13.
Взаимодействие диазокетонов 3Т1-1,2 (находящихся в триплетном возбужденном состоянии и, таким образом, обладающих радикальными свойствами) с тетрагидрофураном в результате типичной для радикалов схемы отрыв-рекомбинация приводит к образованию в качестве конечных продуктов фотореакции гидразонов 9а,11а.
Существенным аргументом в пользу предполагаемой схемы образования продуктов С-Н внедрения 9а,11а из триплетных диазокетонов 3Т1-1,2 являются эксперименты по фотолизу диазокетона 2а в атмосфере кислорода. Известно, что кислород способен эффективно содействовать интеркомбинационной конверсии синглетных возбужденных состояний в триплеты [51]. Таким образом, можно считать, что присутствующий в реакционной смеси кислород способствует переходу синглетных диазокетонов 1S1-1,2 в более низкие по энергии триплетном состояния 3Т1-1,2, увеличивая концентрацию бирадикалов в реакционной среде и, тем самым, способствуя повышению химических выходов продуктов С-Н внедрения 9а, 11а.
В связи с вышесказанным увеличение выхода продукта внедрения 9а при фотолизе диазокетона 2а в атмосфере кислорода может рассматриваться как свидетельство триплетной природы диазокетона 2а (или иного предшественника, ответственного за этот процесс). В препаративном плане, т.е. для увеличения выходов гидразонов типа 9, 11, по-видимому, целесообразно проводить фотолиз диазокетонов 1а, 2а в атмосфере кислорода.
Нельзя также исключить, что проведение в атмосфере кислорода фотолиза тетраалкилзамещенных диазокетонов, у которых в обычных условиях фотолиза наблюдается только перегруппировка Вольфа, приведет, по крайней мере частично, к превращению первоначально образующихся синглетных диазокетонов в триплетные и к образованию по аналогичной схеме тетралкилзамещенных продуктов С-Н внедрения.
В Ы В О Д Ы
1. На примере трех пар региоизомеров детально разработана методика получения 5,5-диарил-2,2-диметил- и 2,2-диарил-5,5-диметилзамещенных диазокетонов ряда тетрагидрофурана с помощью 3-х- и 5-ти-стадийного синтеза из ацетилена Фаворского и пара-замещенных бензофенонов, которая дает возможность синтезировать эти диазосоединения с общими выходами 30-58 и 35-66% соответственно.
2. Термолиз 5,5-ди(пара-фторфенил)-2,2-диметилзамещенных диазокетонов ряда ТГФ в растворе тетрагидрофурана, диоксана и в твердой фазе при температурах 65-100 оС приводит к образованию относительно инертных в этих условиях 2,2-диметил-4,4-диарилкетенов оксетанового ряда.
3. Региоизомерные 2,2- и 5,5-ди(пара-фторфенил)замещенные диазокетоны ряда ТГФ при коротковолновом облучении (λ > 210 нм) претерпевают практически одни и те же фотохимические превращения, и в этих условиях фотолиза различия в их структуре не сказываются на направлении и соотношении наблюдаемых фотопроцессов.
4. Образование продуктов С-Н-внедрения при фотолизе 2,2- и 5,5-ди(пара-фторфенил)замещенных диазокетонов тетрагидрофуранового ряда в растворе ТГФ, по-видимому, происходит из триплетного возбужденного состояния диазосоединения.
Список литературы (к обсуждению):
41. Ando, W. Photochemistry of Diazocompounds. In: The Chemistry of Diazonium and Diazo Groups; Patai, S., Ed. Wiley: Chichester, 1978, P. 1, 341.
42. Regitz M., Maas G. Diazo Compounds. Properties and Synthesis. Acad. Press, New York, 1986, 166; Zollinger H. Diazo Chemistry II, VCH Verlagsgesellshaft, Weinheim. 1995, 305.
43. Bertrand G., Ed. Carbene Chemistry. Marcel Dekker, Inc: New York. 2002, 301 p.
44. Platz M. S., Ed. Kinetics and Spectroscopy of Carbenes and Biradicals. Plenum Press, New York, 1990, 372 p.;