

4. Болезнь Кеннеди или Спино-бульбарная мышечная атрофия.
Болезнь Кеннеди впервые была описана У. Кеннеди в 1968г. Болезнь Кеннеди проявляется в возрасте 40 лет и старше, характеризуется медленным прогрессированием, поражением мимических мышц и бульбарных ядер черепно-мозговых нервов. Клиническая картина представлена атрофией мышц, особенно проксимальных отделов нижних и верхних конечностей, интен-ционным тремором, фасцикуляциями мускулатуры лица.
Болезнь Кеннеди наследуется по рецессивно сцепленному с Х-хромосомой типу. Ген этого заболевания располагается в области хромосомы Хq13.
Дебют заболевания характеризуется слабостью и атрофией мускулатуры проксимальных отделов верхних конечностей, ограничением объема движений в руках, спонтанными фасцикуляция-ми, угнетением сухожильных рефлексов с двуглавой и трехглавой мышц плеча.
Следующим этапом развития заболевания является проявление бульбарных расстройств: атрофия языка, фибрилляция языка, дизартрия, поперхивание. На более поздних стадиях болезни присоединяется атрофия мышц проксимальных отделов нижних конечностей, атрофия мышц тазового пояса. Больным становится тяжело вставать, ходить, подниматься по лестнице. Возникают псевдогипертрофии икроножных мышц, наблюдается гинекомастия.
При биохимическом исследовании крови выявляется умеренное увеличение активности креатинфосфокиназы.
Электромиографическое исследование показывает поражение передних рогов спинного мозга.
При световой микроскопии видны мелкие и крупные участки атрофированных мышечных волокон, увеличение количества гипертрофированных мышечных волокон, снижение количества нормальных волокон.
При электронной микроскопии регистрируется диффузное изменение миофибрилл, 2-полос, скопление ядер миоцитов, фрагментация саркоплазматической сети.
Критерии постановки диагноза болезни Кеннеди:
1)Х-сцепленный рецессивный тип наследования;
2)начало заболевания в период от 40 до 60 лет;
3)слабость и атрофии мышц сначала верхних, затем нижних конечностей;
4)псевдогипертрофии икроножных мышц;
5)наличие бульбарных симптомов: атрофия языка, дизартрия, дисфония, поперхивание и другие;
6)развитие гинекомастии;
7)в биоптатах скелетной мускулатуры гипертрофированные и атрофированные мышечные волокна;
8)при электромиографическом исследовании присутствуют признаки денервации;
9)течение медленно прогрессирующее.
Дифференциальную диагностику следует проводить с другими видами спинальных мышечных атрофий, бокового мышечного атрофического склероза, псевдогипертрофических форм прогрессирующих мышечных дистрофий.
Дополнительный материал.
Вид патологии. |
Локализация гена. |
Тринуклеотидный повтор. |
Число тринуклеотидных повторов. |
|
норма |
патология |
|||
Синдром ломкой Х-хромосомы (FRAXA) |
Xq 27,3 |
ЦГГ |
5—50 |
>200 |
Спино-бульбарная мышечная атрофия |
Xq 11-12 |
ЦАГ |
17—26 |
40—52 |
Миотоническая дистрофия |
19q 13,3 |
ЦТГ |
5—27 |
50—1600 |
Хорея Гентинггона |
4р 16,3 |
ЦАГ |
11—34 |
>42 |
Рис.1. Синдром ломкой Х хромосомы. Синдром Мартина-Белл.
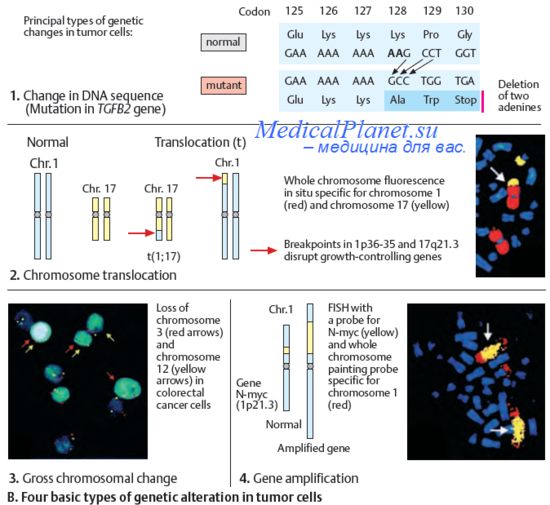
Рис.2. Хорея Гентингтона. Признаки и причины хореи Гентингтона.
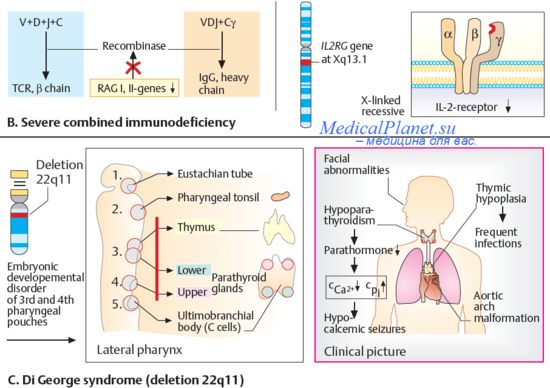
Рис.3. Миотоническая дистрофия Куршмана-Штейнерта-Баттена. Признаки и причины миотонической дистрофии Куршмана-Штейнерта-Баттена.

Литература.
http://medicalplanet.su/genetica/197.html
http://medicalplanet.su/genetica/200.html
http://medicalplanet.su/genetica/198.html
http://medicalplanet.su/genetica/199.html
http://www.astromeridian.ru/medicina/3/1862.html