

47. Теория переходного состояния и вывод основного уравнения.
В модели активного комплекса предполагается, что исходная конфигурация реагирующих частиц, обладающих достаточной энергией для преодоления барьера реакции, преобразуется при непрерывном изменении межатомных расстояний и соответствующей энергии системы в активный комплекс, который обязательно превращается в продукты реакции:
АА + В → {ААВ}
 Для
превращения исходных молекул АА и В в
продукты они должны образовать активный
комплекс {ААВ}, в нем старые связи
разрыхлены и наметились новые, но их
конфигурация и энергия не соответствуют
стабильным молекулам.
Для
превращения исходных молекул АА и В в
продукты они должны образовать активный
комплекс {ААВ}, в нем старые связи
разрыхлены и наметились новые, но их
конфигурация и энергия не соответствуют
стабильным молекулам.
E1 и Е2 – энергия активации прямой и обратной реакций
∆Н = Е1-Е – теплота реакции
Вначале АА и В поднимаются до точки М, которая представляет собой энергетический перевал, затем активный комплекс распадается и молекулы А и АВ «скатываются вниз» в потенциальную яму.
Активный комплекс находится в состоянии равновесия с исходными веществами, причем одна из колебательных степеней свободы вырождается в поступательную по координатам реакции и дальше активный комплекс необратимо превращается в продукты. Прямой и обратный процессы с образованием активного комплекса протекают независимо друг от друга.
Скорость реакции согласно теории будет равна числу активных комплексов, проходящих через потенциальный барьер вдоль координаты реакции в единицу времени, в еденице реакционного пространства.
В соответствии с теорией, скорость реакции будет равна числу активированных комплексов, проходящих через потенциальный барьер вдоль координаты реакции в единицу времени, в единице реакционного пространства. Следует также учесть вероятность прохода через барьер со скоростью u*, введя так называемый трансмиссионный коэффициент χ:
r = χCК*/τ или r = χCК*u*/δ (3.10)
С другой стороны, скорость бимолекулярной реакции можно выразить, используя 1й постулат кинетики:
r = kCАCВ (3.11)
или, исключая скорость из уравнений 10 и 11, можно найти величину константы:
![]() (3.12)
(3.12)
В этом уравнении величины u*,δ и CК* нельзя определить экспериментально, но можно выразить через определяемые величины, используя аппарат статистической термодинамики для равновесных процессов. Так, концентрацию активированного комплекса находим из условия равновесия:
CК* = К*САCВ (3.13)
и получим выражение
k = χu*δ-1К* (3.14)
Скорость прохода активированного комплекс через барьер, выразим согласно молекулярно-кинетической теории:
![]() (3.15)
(3.15)
Выделим поступательную сумму по состояниям из выражения константы равновесия
QП*= δh-1(2πm*RT/NA)1/2, (3.16)
где h – постоянная Планка, и определим константу равновесия КС* без одной поступательной степени свободы как
Кс* = К*/QП* (3.17)
Подставим (3.15) - (3.17) в (3.14) и получим искомое выражение для константы скорости:
k =χRTh-1NA-1Кс* (3.18)
Для уточнения физического смысла используемых ранее терминов энергии активации, предэкспоненциального множителя k0, и стерического фактора преобразуем (3.18), выразив константу равновесия через энергию Гиббса
![]() ,
а энергию Гиббса - в виде
,
а энергию Гиббса - в виде
∆Go* = ∆Ho* - T∆So*
![]() (3.19)
(3.19)
Это – основное уравнение теории активированного комплекса.
Из уравнения (19) видно, что величина предэкспоненциального множителя
k0=χRTh-1NA-1.exp(∆So*/R) (3.20)
зависит от частоты колебаний активированного комплекса и изменения энтропии при его образовании.
Для нахождения связи между энергией активации и теплотой активации АК (∆Ho*) прологарифмируем (3.18)
![]() и возьмем производную по температуре:
и возьмем производную по температуре:
![]() (3.21)
(3.21)
Как видно из (3.21) энергия активации реакции Е равна сумме энтальпии активации активированного комплекса и RТ:
Е = ∆Ho*+ RТ (3.22)
Уравнение теории АК можно использовать для расчета предэкспоненциального множителя k0 в случае простых реакций (или оценки для сложных реакций).
Рассмотрим простой случай мономолекулярной реакции:
А A* B
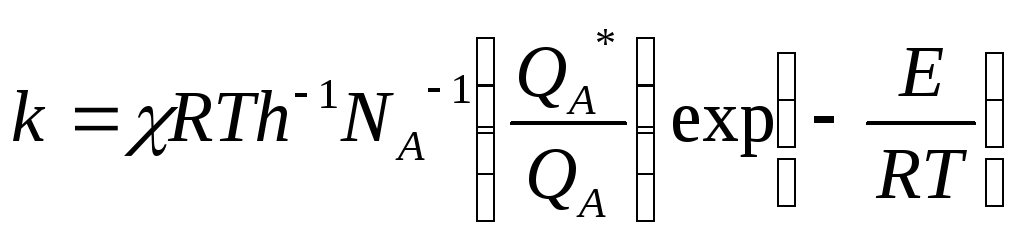 (3.23)
(3.23)
Поскольку суммы по состояниям для А и А* различаются на одну колебательную степень свободы, их отношение равно:
![]() (3.24)
(3.24)
величина
![]() <<
1 при Т > 1000K, и в этой области температур
<<
1 при Т > 1000K, и в этой области температур
![]() (3.25)
(3.25)
подставляя (3.24) в (3.23) получим:
![]() (3.26)
(3.26)
Откуда видно, что k0 = χν, т.е. предэкспоненциальный множитель по порядку величины равен частоте колебаний разрываемой связи. В табл. 4 приведены значения предэкспоненциального множителя типичных мономолекулярных реакций.
48. Связь константы скорости реакции с энтальпией и энтропией активации.
AB
+ С
![]() A…B…C
A…B…C![]() A
+ BC
A
+ BC
Выражение константы скорости для бимолекулярной реакции:
![]() (5),
где χ – трансмиссионный коэффициент,
определяет долю активных комплексов,
прошедших через точку М.
(5),
где χ – трансмиссионный коэффициент,
определяет долю активных комплексов,
прошедших через точку М.
Изменение энергии Гиббса для активных комплексов при стандартных условиях равно теплоте и энтропии активации или константе равновесия Кр#.
![]() ;
;
![]() =>
=>
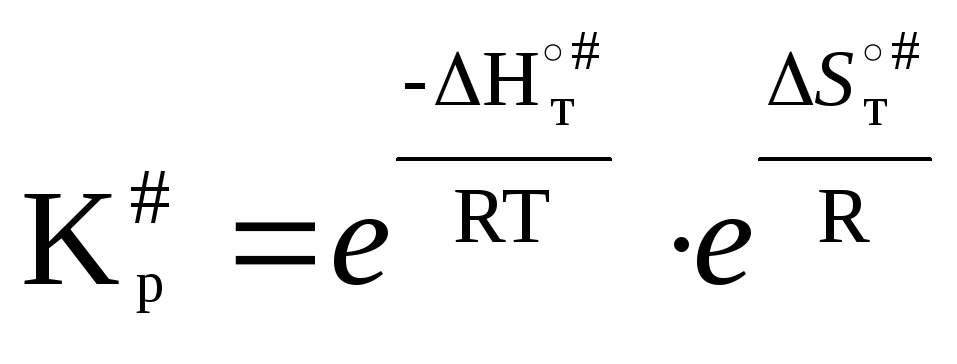 (6)
(6)
Подставив (5) в (6):
 (7)
(7)
Используя уравнение Аррениуса: K1 = K01exp(-E/RT) и (7) можно найти связь между теплотой активации и энергией активации и выражение для K01:
Логарифмируем
(7):
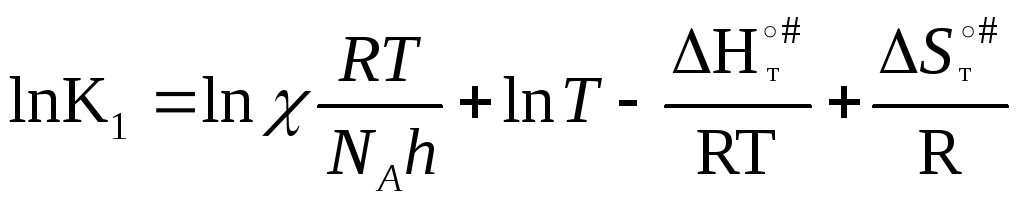
и дифференцируем по Т (∆S0#=const)
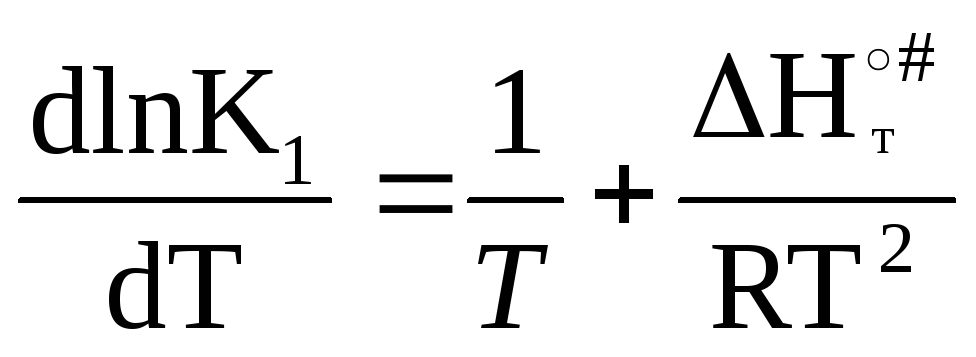 ;
;
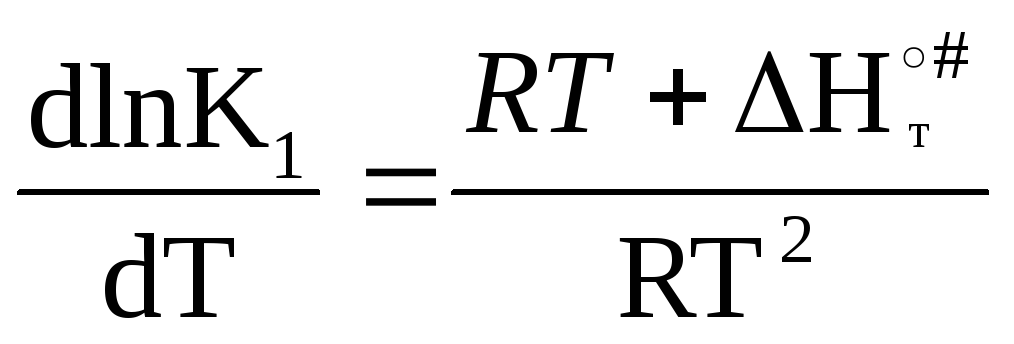 (8)
(8)
Сравнивая с уравнением Аррениуса:
![]() =>
=>
![]() ;
;
![]() (9)
(9)
Подставляя (9) в (7):
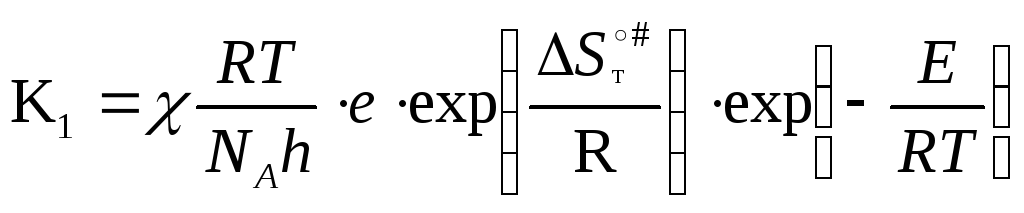 =>
=>
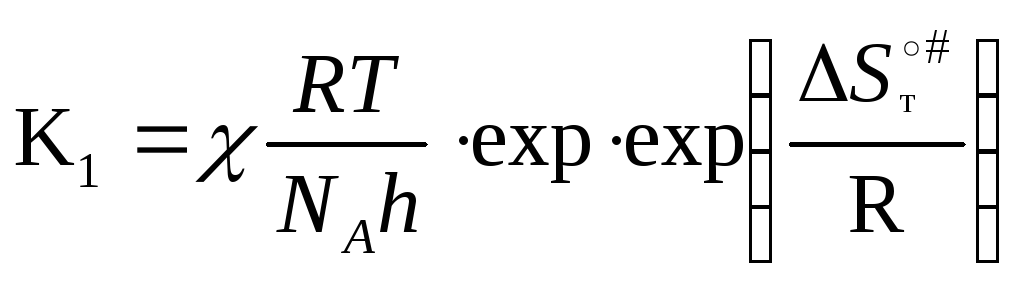
По
величине
![]() реакции делят на «нормальные» (
реакции делят на «нормальные» (![]() =0,
exp(
=0,
exp(![]() /R)=1),
«быстрые» (
/R)=1),
«быстрые» (![]() >>0,
exp(
>>0,
exp(![]() /R)>>1)
и «медленные» (
/R)>>1)
и «медленные» (![]() <<0,
exp(
<<0,
exp(![]() /R)<<1).
/R)<<1).
exp(![]() /R)
приобретает смысл стерического
множителя.
/R)
приобретает смысл стерического
множителя.
49.Цепные р-ии, основные понятия, основные стадии и типы р-ий.
Имеется класс специфических реакций, протекающих в газовой (а также в жидкой фазах) через образование высокоэнергетических возбужденных частиц, радикалов, атомов и др. Здесь термин высокоэнергетический означает, что энергия частиц (Е) больше энергии активации, Еа. Далее эти активные частицы быстро взаимодействуют с исходными реагентами, с образованием продуктов и регенерацией активных частиц. Таким образом, в результате зарождения малого числа активных частиц превращается большое число молекул реагентов. Такой циклический процесс называется цепным, причем повторяющаяся группа реакций называется цепью. Такие процессы впервые были обнаружены Боденштейном (1913), а их интерпретация предложена Нернстом (1916). В последующем было показано, что по цепному механизму протекают многие процессы, такие как окисление углеводородов и других субстратов, крекинг и пиролиз, полимеризация олефинов, галогенирование и т.п.
В цепном процессе выделяют этапы:
1) Зарождение (инициирование) цепи с образованием активных частиц (АЧ).
2) Развитие (продолжение) цепи, характеризуемое длиной цепи, lц, которая представляет собой число молекул реагента, прореагировавших на одну активную частицу, образовавшуюся на этапе зарождения цепи.
3) Обрыв цепи в результате процессов дезактивации/гибели активных частиц, например, на стенке (при малых давлениях) или при тройных соударениях с молекулами реагентов или примесей. Здесь материал стенки или третья частица играют роль «приемника» избыточной энергии, выделяющейся при ассоциации атомов и/или радикалов. За счет передачи этой энергии возможна стабилизация образующихся молекул (Н2 или С12). Кроме указанных реакций, обрыв цепи может происходить за счет взаимодействия активных частиц с добавками (ингибиторами), при котором образуются относительно стабильные радикалы (например, типа стерически затрудненных фенолов или аминов).
Проиллюстрируем эти этапы на примере реакции Н2 + С12= 2НС1:
1)
![]() (инициирование)
(инициирование)
2а)
![]() (продолжение
цепи)
(продолжение
цепи)
2б)
![]() и т.д.
и т.д.
3а)
![]() (обрыв цепи на стенке 3а)
(обрыв цепи на стенке 3а)
3б)
![]() (и в объеме 3б))
(и в объеме 3б))
где М – материал стенки.
Основными признаками наличия цепного процесса являются:
существенное увеличение скорости развитого процесса (r) по сравнению с начальной скоростью (ri); отношение r/ri равно средней длине lц.
образование нескольких молекул продукта на 1 молекулу инициатора или на 1 квант инициирующего излучения,
зависимость скорости реакции от материала стенки реактора и отношения поверхности к объему реакционного пространства,
сильная зависимость скорости реакции от концентрации добавок ингибиторов.
Наиболее трудным этапом цепного процесса обычно является инициирование. Этому этапу соответствует индукционный период наблюдаемой кинетической кривой. Цепной процесс может быть инициирован соответствующим излучением или веществами-инициаторами, например, из класса пероксидов, диазосоединений и т.п. По мере увеличения концентрации активных частиц (в рассмотренном примере радикалов H и Cl) скорость реакции существенно возрастает до стационарного значения (в случае неразветвленной цепи) или до выгорания/взрыва (в случае разветвленной цепи) реакционной смеси. На заключительном этапе реакции, когда процессы обрыва АЧ преобладают над инициированием цепи, скорость реакции убывает во времени (рис. 17).
|
3 2
1 |
Рис. 18. Изменение скорости цепной реакции во времени на этапах: 1- инициирование цепи; 2- развитие; 3- обрыв цепи |
|
Время реакции, t |
Различают неразветвленные и разветвленные цепные реакции. Процесс называется неразветвленным, когда на этапе продолжения цепи превращение одной активной частицы приводит к образованию одной новой АЧ. Если при этом образуется больше одной новой активной частицы, такой цепной процесс называется разветвленным.
|
|
|
|
 r
r
 2
2
