

Журнал неврологии и психиатрии / 2006 / NEV_2006_01_01
.pdfОБЩИЕ ВОПРОСЫ НЕВРОЛОГИИ И ПСИХИАТРИИ
Полиморфизм генов системы детоксикации и предрасположенность к болезни двигательного нейрона в российской популяции
В.И. СКВОРЦОВА, П.А. СЛОМИНСКИЙ, М.И. ШАДРИНА, Г.Н. ЛЕВИЦКИЙ, Н.И. ЛЕВИЦКАЯ, А.В. АЛЕХИН, А.Л. ЖЕРЕБЦОВА, А.В. СЕРДЮК, С.А. ЛИМБОРСКАЯ
Detoxication gene polymorphism and susceptibility to sporadic motor neuron disease in Russian population
V.I. SKVORTSOVA, P.A. SLOMINSKY, M.I.SHADRINA, G.N. LEVITSKY, N.I. LEVITSKAYA, A.V. ALEKHIN, A.L. ZHEREBTSOVA, A.V. SERDYUK, S.A. LIMBORSKA
Кафедра фундаментальной и клинической неврологии Российского государственного медицинского университета, Институт молекулярной генетики РАН, Москва
Болезнь двигательного нейрона (БДН) — это селективная дегенерация мотонейронов коры головного мозга, ствола и спинного мозга. В ее патогенезе могут быть задействованы многие генетические системы. Одним из патогенетических факторов БДН является свободнорадикальное повреждение клеток. Важным метаболическим процессом, в ходе которого могут образовываться свободные радикалы, впоследствии преобразующиеся в нетоксичные продукты, является детоксикация. Основными участниками данного процесса являются цитохромы Р-450 (CYP2E1, CYP2D6), глутатион-S-трансферазы (GSTM1, GSTТ1, GSTP1) и N-ацетилтрансферазы (NAT2). С целью уточнения роли генов системы детоксикации в развитии БДН был изучен полиморфизм этих генов у 72 больных из Москвы и в контрольной группе лиц, проживающих в России. Выявлены достоверные различия в частотах аллелей CYP2E1*1D, CYP2D6*4 и гомозигот по генотипу GSTM1(0/0) между больными и контролем. Анализ полиморфизма генов GSTT1, GSTP1 и NAT2 не обнаружил различий в распределении генотипов и аллелей между основной и контрольной группами. Установлено, что генотип GSTР1*A/GSTP1*A ассоциирован с классическим вариантом БДН (равномерным поражением центральных и периферических мотонейронов), а аллель GSTР1*B — с ее сегментарно-ядерным и пирамидным вариантами.
Ключевые слова: болезнь двигательного нейрона, молекулярно-генетическое исследование, гены системы детоксикации.
Motor neuron disease (MND) is caused by selective degeneration of motor neurons of the cerebral cortex, brain stem and spinal cord. Many genetic systems are thought to be involved in pathogenesis of this complex disease. A significant etiological factor of MND may be oxygen free radicals, which damage neuronal cells when they are present in high concentrations. Detoxication processes resulting in the formation of free radicals, which subsequently transformed into nontoxic products, are also critical for the disease development. The major participants of these processes are cytochromes P-450 (CYP2E1, CYP2D6), glutathione–S-transferases (GSTM1, GSTT1, GSTP1) and N-acetyltransferases (NAT2). To investigate a role of genes of detoxication system in development of MND, we study polymorphisms in these genes in 72 patients with MND from Moscow and controls from Russia. Significant statistical differences have been found in frequency of the alleles CYP2E1*1D, CYP2D6*4 and GSTM1(0/0) and genotypes homozygous for GSTM1
(0) between the study and control groups. The analysis of GSTT1, GSTP1 and NAT1 gene polymorphisms has revealed no between-group differences in distribution of different alleles and genotypes. The GSTP1*A/ GSTP1*A genotype was associated with a classical upper and lower motor neuron involvement and the GSTP1*A allele with predominant lower and upper motor neuron involvement.
Key words: motor neuron disease, molecular genetic study, detoxication system genes.
Болезнь двигательного нейрона (БДН) — это нейроде- |
на 100 000 населения. По мере увеличения возраста БДН |
|
генеративное заболевание с началом в позднем возрасте, |
|
встречается чаще [42]. Больные обычно погибают в течение |
которое характеризуется прогрессирующей дегенерацией |
2—5 лет от начала болезни [32]. |
|
мотонейронов головного и спинного мозга. Частота встре- |
|
Наиболее частой формой БДН является боковой амио- |
чаемости БДН в развитых странах составляет до 5 случаев |
трофический склероз (БАС) — мультифакторное заболева- |
|
|
|
ние, вызываемое сложным взаимодействием наследствен- |
|
|
ных факторов и факторов окружающей среды [36]. По дан- |
© Коллектив авторов, 2006 |
|
ным эпидемиологических исследований, в разных популя- |
|
циях семейная форма БДН встречается в 5—10% случаев. |
|
|
|
|
Zh Nevrol Psikhiatr Im SS Korsakova 2006;106: 1: 4—13 |
Клинические и патоморфологические черты семейной и |
|
4 |
|
ЖУРНАЛ НЕВРОЛОГИИ И ПСИХИАТРИИ, 1, 2006 |
Nv_1_06_.p65 |
4 |
20.12.2005, 10:38 |
ГЕНЕТИКА БОЛЕЗНИ ДВИГАТЕЛЬНОГО НЕЙРОНА
спорадической БДН идентичны. Несмотря на многолетние исследования, генетическая природа БДН до конца не уточ- нена.
Важным элементом патогенеза БДН является повышенная продукция свободных радикалов, которые повреждают мотонейроны [35]. Известно, что важную роль в поддержании нормального уровня свободных радикалов играет супероксиддисмутаза (СОД-1) [44]. Мутации в гене СОД-1 обнаружены в 20% случаев семейной БДН и в небольшом проценте спорадических случаев [5, 6]. Мутации СОД-1 обнаружены и у больных в Российской Федерации [2].
Свободные радикалы, в частности, образуются в ходе процессов детоксикации, впоследствии преобразуясь в нетоксичные продукты. Детоксикация играет ключевую роль в метаболизме ксенобиотиков, в том числе большинства препаратов и токсичных соединений окружающей среды [9], из которых многие могут принимать участие в дегенерации нейронов. Пути детоксикации ксенобиотиков включают их инактивацию в фазе I и инактивацию высокотоксичных промежуточных метаболитов в фазе II. Суперсемейство генов цитохромов-450 (CYP) (фаза I) и глутатион-S-транс- фераз, а также ариламин-N-ацетилтрансферазы 2 (NAT2) (фаза II) играют основную роль в этих процессах биотрансформации.
Цитохромы (CYPs) представляют суперсемейство ферментов, отвечающих за окисление, перекисное окисление и восстановление эндогенных и экзогенных веществ. Семейство цитохромов CYP1—3 активно метаболизирует широкий спектр ксенобиотиков и играет важную роль в защите организма от их воздействия [21]. Изменения активности CYP могут привести к усилению индивидуальной восприимчивости к действию как эндогенных, так и экзогенных токсинов. Некоторые цитохромы, такие как CYP2E1, CYP2D6, CYP2A1 и CYP1A2, могут участвовать в патогенезе неврологических заболеваний [11, 17, 51, 56].
Цитохром CYP2E1 (этанолиндуцибельный фермент) катализирует окисление более 75 ксенобиотиков, в том числе этанол, лекарственные препараты и некоторые потенциальные канцерогены [33]. CYP2E1 обладает уникальным свойством преобразовывать многие ксенобиотики в их токсич- ные метаболиты; часто это свободные радикалы, которые предположительно задействованы в патогенезе БДН [38]. Недавно был описан инсерционный полиморфизм размером 96 пар оснований (п.о.) — CYP2E1*1D в промоторе гена CYP2E1, локализующийся между –2270 и –1672 позициями [35]. Частота этого полиморфизма в этнических группах колеблется от 2% у европеоидов до 10% у американских негров [18]. Он часто наблюдается у детей, страдающих перинатальной патологией, связанной с алкоголизмом матери [39]. Инсерционный полиморфизм CYP2E1 ассоциирован с повышенным риском развития инфильтративного туберкулеза легких [10]. Генотипы, содержащие инсерцию, ассоциированы с превышением нормы активности фермента [38]. Такое возрастание индуцированной активности фермента, вероятно, стимулирует образование свободных радикалов и нейротоксичных метаболитов. Поэтому можно предположить, что данный полиморфизм ассоциирован с повышенным риском развития БДН.
Другим цитохромом, который может участвовать в различных патологических процессах, является дебризокин-4- гидроксилаза (CYP2D6). Она метаболизирует широкий спектр ксенобиотиков (лекарственных препаратов, металлов, а также химикатов, образующихся в природе или на производствах). Примерно у 5—10% европеоидов отмечается снижение функции этого фермента — фенотип «медленной метаболизации» (MM) [29]. Этот фенотип ассоциирован с широким спектром заболеваний, включая рак [62], болезни Паркинсона [54] и Альцгеймера [13]. Существует по крайней мере 30 дефектных аллелей CYP2D6, из которых 6 лежат в основе 95—99% фенотипов MM [29]. Одним из наиболее полно охарактеризованных вариантов является аллель
CYP2D6*4, возникающий в результате замены гуанина на аденин на границе 3-го интрона и 4-го экзона гена [22]. Гомозиготность по аллелю CYP2D6*4 лежит в основе 75% фенотипов ММ [53]. Из нескольких исследований по изуче- нию ассоциации полиморфизма CYP2D6*4 с развитием БДН
âодном [30] частота встречаемости гомозигот по CYP2D6*4 среди больных с БДН не отличалась от контроля, тогда как
âдругом [56] была повышена, а частота фенотипа ММ повышенной не была, и это дало основание полагать, что наличие аллеля CYP2D6*4 может быть фактором риска развития БДН.
Метаболиты фазы I детоксикации, образуемые цитохромами, часто потенциально более вредны, чем исходные вещества, и важно, чтобы они не накапливались. Ферменты фазы II инактивируют эти промежуточные метаболиты, катализируя их связывание с кофакторами, которые превращают их в гидрофильные формы, что облегчает выведение таких метаболитов [48]. Наиболее важными участниками фазы II являются глутатион-S-трансферазы (GSTs) — полигенное суперсемейство изоферментов, широко представленное в животном мире [37]. Первичной их функцией является детоксикация, которая опосредована конъюгацией большого количества электрофильных соединений с восстановленным глутатионом (GSH) [47]. У человека обнаружены различные изоферменты GST, ряд которых обладает
тканеспецифичной экспрессией [61]. Известно по крайней мере 7 семейств растворимых GSTs: α, µ, π1, σ, θ, κ, ξ [31]. Отдельные GSTs полиморфны, и некоторые аллельные ва-
рианты их генов обусловливают снижение активности ферментов. Глутатион-S-трансферазы θ1 (GSTT1), µ1 (GSTM1) и π1 (GSTP1) представляют наибольший интерес. Известны делеционные, или «нулевые», аллели генов GSTT1 и GSTM1, которые определяют невозможность экспрессировать белок [55]. Они встречаются часто — «нулевые» генотипы GSTT1 и GSTM1 (0/0) имеются у 10—20 и 40—65% европеоидов соответственно [50]. Ген GSTP1 имеет несколько полиморфизмов. Функционально значимой является замена аденина на
гуанин в 5-м экзоне гена, которая приводит к замене изолейцина на валин в 105-м кодоне (105 Ile→Val). Она обусловливает изменение термоустойчивости и специфической активности валинсодержащей изоформы [24, 65]. Считают, что эти варианты GST увеличивают восприимчивость человека к различным заболеваниям, в том числе к хроническому бронхиту, разным видам рака [26] и к неврологической патологии [59].
Другим важным участником фазы II является ариламин- N-ацетилтрансфераза типа 2 (NAT2), катализирующая N-ацетилирование ксенобиотиков с первичной ароматиче- ской или гидразиновой структурой [27], например токсич- ные нитрозамины табачного дыма, антиоксиданты и пестициды. NAT2 участвует в метаболизме лекарств, в том числе в лекарственных взаимодействиях [9, 57]. Способность NAT2 N-ацетилировать различные ксенобиотики связана с полиморфизмом гена NAT2. В зависимости от вариантов активности последнего всех людей можно разделить на две группы — медленных (МА) и быстрых (БА) ацетиляторов. МА гомозиготны по рецессивным формам гена NAT2, имеют два «медленных» аллеля, и уровень экспрессии белка NAT2 у них снижен на 20%. БА имеют по крайней мере один из «быстрых» аллелей NAT2 дикого типа [14]. Примерно 50% европеоидов являются МА, но представленность БА и МА различна в зависимости от географического региона [15]. Проведен ряд исследований, в ходе которых установлено, что полиморфизмы ацетилирования ассоциированы с развитием различных заболеваний, в частности некоторых видов рака [4, 28, 34, 64] и неврологических, например болезни Паркинсона [7].
Эти данные, а также тот факт, что у больных с хрони- ческими неврологическими заболеваниями, по-видимому, имеет место снижение способности детоксицировать определенные экзогенные соединения [58, 62], дают основание
ЖУРНАЛ НЕВРОЛОГИИ И ПСИХИАТРИИ, 1, 2006 |
5 |
Nv_1_06_.p65 |
5 |
20.12.2005, 10:38 |

ОБЩИЕ ВОПРОСЫ НЕВРОЛОГИИ И ПСИХИАТРИИ
полагать, что гены системы детоксикации могут быть косвенно вовлечены в патогенез БДН.
Цель настоящего исследования состояла в проверке высказанной гипотезы, согласно которой гены системы детоксикации вовлекаются в патогенез БДН. Нами проведен сравнительный анализ распределения вариантов генотипов CYP2E1, CYP2D6, GSTT1, GSTM1, GSTP1 и NAT2 у пациентов с БДН и в контрольной группе.
Материал и методы
После информированного согласия пациентов образцы крови были взяты у 75 проживающих в Российской Федерации (средний возраст 55,5±10,9 года), у которых предполагалось наличие спорадической БДН.
Больные были обследованы на кафедре фундаментальной и клинической неврологии РГМУ. В соответствии с классификацией F. Norris и соавт. [46] их разделили на группы БАС и прогрессирующего бульбарного паралича (ПБП) (табл. 1).
Типы прогрессирования оценивали по шкале F. Norris и соавт. [45] в ходе динамического наблюдения каждые 6 мес вплоть до смерти больного. Пациенты с быстрым прогрессированием теряли за 6 мес более 10 баллов, со средним прогрессированием — от 5 до 10 баллов, с медленным — менее 5 баллов. Согласно данным регрессионного анализа, быстрый, средний и медленный типы прогрессирования достоверно различались: соответственно r=–0,96, p<0,001; r=–0,97, p<0,001; r=–0,98, p<0,001.
Контрольную группу составили 105 жителей Москвы, не находившихся между собой в родстве; она была сопоставима по полу, возрасту и национальности с основной группой.
Таблица 1. Клинические характеристики больных с БДН
Характеристика |
Kоличество больных |
||
|
|
||
àáñ. |
% |
||
|
|||
|
|
|
|
Форма/дебют: |
|
|
|
шейный дебют БАС |
37 |
49,3 |
|
поясничный дебют БАС ALS |
13 |
17,3 |
|
грудной дебют БАС |
5 |
6,7 |
|
диффузный дебют БАС |
4 |
5,3 |
|
ÏÁÏ |
16 |
21,3 |
|
Вариант: |
|
|
|
классический |
40 |
53,3 |
|
сегментарно-ядерный |
23 |
30,7 |
|
пирамидный |
12 |
16,0 |
|
Тип прогрессирования: |
|
|
|
быстрый |
41 |
54,6 |
|
средний |
17 |
22,7 |
|
медленный |
17 |
22,7 |
|
|
|
|
|
6
ДНК выделяли из лейкоцитов стандартными методами [40].
Генотипирование CYP2E1. Наличие инсерции 96 п.о. в промоторе гена CYP2E1 устанавливали с помощью полимеразной цепной реакции (ПЦР) [18]. Использовали праймеры 5'-GTGATGGAAGCCTGAAGAACA-3' и 5'-CTTTGGT GGGGTGAGAACAG-3'. ПЦР проводили в 20 мкл реакционной смеси, содержащей 5 пмоль каждого праймера, 2 мкл 10½ПЦР буфера (500 ммоль трис-НСl, рН 8,8, 150 ммоль (NH4)2SO4, 50 ммоль MgCl2, 2 мг/мл альбумина телячьей сыворотки), 1,0 ммоль каждого динуклеотидтрифосфата (дНТФ) и 0,5 ед. Taq ДНК-полимеразы (MBI Fermentas). Условия ПЦР: денатурация при 95°С в течение 5 мин, далее 30 циклов при 95°С в течение 1 мин, при 66°С в течение 1 мин и при 72°С в течение 1 мин. Продукты ПЦР растворяли в 8% полиакриламидном геле, окрашенном бромидом этидия. Длина фрагмента, соответствующего аллелю без инсерции и содержащего 6 повторов в области 5'-конца (CYP2E1*1C), составляла 633 п.о., длина фрагмента, соответствующего аллелю с инсерцией и содержащего 8 повторов в области 5'-конца (CYP2E1*1D), — 729 п.о.
Генотипирование СYР2D6. Применяли метод генотипирования по М. Brown и соавт. [12]. Он позволяет отличить аллели CYP2D6*4 от других аллелей CYP2D6 при помощи ПЦР и рестрикционного теста с использованием рестриктазы BstNI. Этот фермент разрезает аллель дикого типа в положении 1934, но не реагирует с мутантными аллелями. Положение 5' ПЦР-праймера было изменено, чтобы вклю- чить неполиморфный сайт рестрикции BstNI в положении 1772. Смесь для реакции амплификации содержала 5 пмоль каждого праймера (5'-GGTGTTCCTCGCGCGCTATG-3' и 5'- CTCGGTCTCTCGCTCCGCAC-3'), 2 мкл 10½ПЦР-буфера, 1,0 ммоль каждого дНТФ и 0,5 ед. Taq ДНК-полимеразы (MBI Fermentas). Условия ПЦР: денатурация при 95°С в те- чение 5 мин, затем 30 циклов при 95°С в течение 1 мин, при 65°С 1 мин и при 72°С 1 мин. Продукты рестрикции экстрагировали с помощью аликвот ПЦР-продуктов объемом 10 мкл (длиной 421 п.о.) и 1 ед. MvaI (изошизомер BstNI; MBI Fermentas) и инкубировали при 37°С в течение ночи. Продукты рестрикции подвергали электрофорезу в 8% полиакриламидном геле и визуализировали путем окраски бромидом этидия и ультрафиолетового облучения. Гомозигот по CYP2D6*4 диагностировали по наличию двух полос длиной 77 и 344 п.о., а гомозигот — по аллелям, отличным от CYP2D6*4, по наличию трех полос длиной 77, 161 и 183 п.о.
Генотипирование GSTM1 и GSTT1. Гомозиготные делеции генов GSTM1 и GSTT1 определяли с помощью методик множественной ПЦР с праймерами для 4-го экзона гена ГТФ- циклогидролазы-1 (ГЦГ-1) в качестве внутреннего контроля [3]. ПЦР проводили в 20 мкл реакционной смеси, содержащей 2 мкл 10½ПЦР-буфера, 1,0 ммоль каждого дНТФ, 0,5 ед. Таq ДНК-полимеразы (MBI Fermentas) и 5 пмоль каждого праймера (ген GSTM1: 5'-GAACTCCCTGAAAA GCTAAGC-3' и 5'-GTTGGGGCTCAAATATACGGTGG-3'; ген GSTT1: 5'-ТТССТТАСТGGТССТСАСАТСТС-3' и 5'-TCACCGGATCAG GCCAGCA-3'; 4-й экзон гена ГЦГ-1: 5'-GTCCTTTTTGTTTTAT GAGGAAGGC-3' и 5'-GGTGATGCACTCTTATAATCTCAGC-3'). Условия ПЦР: денатурация при 95°С в течение 5 мин, затем 30 циклов при 95°С в течение 1 мин, при 63°С 1 мин и при 72°С 1 мин. Продукты ПЦР анализировали с помощью электрофореза в 8% полиакриламидном геле и визуализировали путем окраски бромидом этидия и ультрафиолетового облучения. Наличие или отсутствие генов GSTT1 и GSTM1 определяли по наличию или отсутствию полос длиной 480 п.о. или 215 п.о. соответственно. Наличие полосы длиной 297 п.о. (4-й экзон гена ГЦГ-1) означало, что амплификация прошла успешно. Данный метод не позволяет различить гетеро- и гомозиготный фенотипы, но лишь убедительно выявляет «нулевые» генотипы.
ЖУРНАЛ НЕВРОЛОГИИ И ПСИХИАТРИИ, 1, 2006
Nv_1_06_.p65 |
6 |
20.12.2005, 10:38 |

Генотипирование GSTP1. Полиморфизм Ile105Val в 5-м экзоне гена GSTP1 выявляли с помощью ПЦР и рестрикционного анализа [63] с праймерами 5'-GTAGTTTGCCCAAGGTCAAG-3' и 5'-AGCCAACCTGAGGGGTAAG-3'. Смесь для реакции амплификации содержала 2 мкл 10½ПЦР-буфера, 1,0 ммоль каждого дНТФ, 5 пмоль каждого праймера и 0,5 ед. Таq ДНКполимеразы (MBI Fermentas). Условия ПЦР: денатурация при 95°С в течение 5 мин, затем 30 циклов при 95°С по 1 мин, при 65°С в течение 1 мин и при 72°С 1 мин. Продукты рестрикции экстрагировали с помощью аликвот ПЦР-про- дуктов объемом 10 мкл и 1,5 ед. Alw26I (изошизомер BsmAI; MBI Fermentas), инкубировали при 37°С в течение ночи. Фрагменты рестрикции растворяли при помощи электрофореза в 8% полиакриламидном геле и визуализировали путем окраски бромидом этидия и ультрафиолетового облучения. Длина фрагмента аллеля GSTP1*А, содержащего изолейцин в 105-м кодоне, составляла 320 п.о., фрагмента аллеля GSTP1*B, содержащего валин в 105-м кодоне, — 222 п.о.
Генотипирование NAT2. Полиморфизм гена NAT2 изуча- ли, как было описано ранее [8, 60]. Выявляли три наиболее распространенных «медленных» аллеля — S1, S2 и S3 (NAT2*5, NAT2*6 и NAT2*7 соответственно) и один «быстрый» аллель дикого типа — F1 (NAT2*4). Амплификацию при ПЦР проводили с праймерами 5'-GCTGGGTCT GGAAGCTCCTC-3' и 5'-TTGGGTGATACATACACAAGGG-3' в 20 мкл реакционной смеси, содержащей 2 мкл 10½ПЦР-буфера, 1,0 ммоль каждого дНТФ, 5 пмоль каждого праймера и 0,5 ед. Таq ДНКполимеразы (MBI Fermentas). Условия ПЦР: денатурация при 95°С в течение 5 мин, затем 35 циклов при 95°С в течение 1 мин, при 57°С 1 мин и при 72°С 1 мин. Продукты амплификации обрабатывали рестриктазами КрnI (для выявления аллеля NAT2*5 [S1]), TaqI (для выявления аллеля NAT2*6 [S2]) и ВаmHI (для выявления аллеля NAT2*7 [S3]). Фрагменты рестрикции растворяли при помощи электрофореза в 8% полиакриламидном геле и визуализировали путем окраски бромидом этидия и ультрафиолетового облучения.
Статистическая обработка. Для анализа таблиц сопряженности применяли Statistica software 6.0 и программу R½C; для определения величины р без погрешности пользовались алгоритмом Метрополиса [49].
У больных и лиц контрольной группы рассчитывали уравнение Харди — Вейнберга для генотипов CYP2E1, CYP2D6, GSTP1 и NAT2, чтобы определить, соответствует
ГЕНЕТИКА БОЛЕЗНИ ДВИГАТЕЛЬНОГО НЕЙРОНА
ли распределение аллелей ожидаемому (генотипы GSTM1 и GSTT1 обозначали как «дикий» или «нулевой», что не позволяло провести прямое вычисление уравнения Харди — Вейнберга). Различия в распределении генотипов анализированных генов у пациентов и лиц контрольной группы оценивали с помощью критерия χ2. Анализ ассоциации между генотипами и клиническими характеристиками проводили с помощью непараметрических ранговых корреляций (γ или Спирмена).
Результаты
Полиморфизм гена CYP2E1. Результаты генотипирования CYP2E1 представлены в табл. 2. Необходимо отметить, что мы выявляли только аллель CYP2E1*1C, несущий 6 повторов в промоторной области, а также инсерционный аллель — CYP2E1*1D, несущий 8 повторов, но не аллель дикого типа CYP2E1*1А, несущий только 5 повторов. Аллель CYP2E1*1D встречался редко, особенно в контрольной группе, и разли- чий в его частоте между контрольной группой (2,5%) и популяцией европеоидов не выявлено (2%) [18]. Однако достоверные различия были установлены между группой контроля и пациентами с БДН. При БДН частота аллеля CYP2E1*1D была достоверно выше (14% при 2,5% в контроле; р<0,001). Анализ распределения генотипов не выявил генотипа CYP2E1*1D/ CYP2E1*1D в контрольной группе. Эти данные согласуются с результатами исследования популяции европеоидов [18]. Сравнительный анализ распределения генотипов выявил достоверные различия между группой больных и контролем в обследованной нами российской популяции (р=0,0018). Отмечалось достоверное увеличение частоты гомозиготных носителей аллеля CYP2E1*1D в группе БДН (см. табл. 2). Распределение генотипов соответствовало уравнению Харди
— Вейнберга. Поскольку генотипы, содержащие инсерцию, приводят к повышению активности фермента [38], мы объединили гетерозиготных носителей аллеля CYP2E1*1D (а) и гомозиготных носителей аллеля CYP2E1*1D (б) в один генотип. Частота комбини-
Таблица 2. Распределение аллелей и генотипов инсерционного полиморфизма гена CYP2E1 у больных с БДН и в контрольной группе
Показатель |
ÁÄÍ |
|
|
Kонтроль |
χ2 |
p |
|
|
|
|
|
||||
n |
% |
n |
% |
||||
|
|
|
|||||
|
|
|
|
|
|
|
|
Частота аллелей |
|
|
|
|
|
|
|
CYP2E1*1C (Ins96(–)) |
129 |
86,0 |
205 |
97,5 |
|
|
|
CYP2E1*1D (Ins96(+)) |
21 |
14,0 |
5 |
2,5 |
7,63 |
<0,001 |
|
Частота генотипов |
|
|
|
|
|
|
|
CYP2E1*1C/ CYP2E1*1C |
60 |
80,0 |
100 |
95,0 |
|
|
|
CYP2E1*1C/ CYP2E1*1D (a) |
11 |
14,7 |
5 |
5,0 |
|
|
|
CYP2E1*1D/ CYP2E1*1D (á) |
4 |
5,3 |
0 |
— |
12,64^ |
0,0018^ |
|
CYP2E1*1C/ CYP2E1*1C |
60 |
80,0 |
100 |
95,0 |
|
|
|
(à+á) |
15 |
20,0 |
5 |
5,0 |
11,6 |
<0,001 |
|
Всего |
75 |
— |
105 |
— |
|
|
|
|
|
||||||
Примечание. Здесь и в табл. 2—4: ^ — χ2 рассчитан при помощи программы R½C; достоверность корреляции при р<0,05. |
|
||||||
ЖУРНАЛ НЕВРОЛОГИИ И ПСИХИАТРИИ, 1, 2006 |
|
|
|
|
7 |
||
Nv_1_06_.p65 |
7 |
20.12.2005, 10:38 |

ОБЩИЕ ВОПРОСЫ НЕВРОЛОГИИ И ПСИХИАТРИИ
рованного генотипа CYP2E1 (а+б) также достоверно преобладала у больных по сравнению с контролем (р<0,001).
Проанализировали ассоциацию аллеля CYP2E1*1D с развитием болезни. Исследовали корреляцию между наличием комбинированного генотипа CYP2E1 и различными клиническими характеристиками, приведенными в табл. 1. Корреляция была установлена только между формой/дебютом БДН и наличием комбинированного генотипа (γ-корреляция: r=2,838, p=0,005; корреляция Спирмена: r=0,4, p=0,033). Дальнейший анализ показал, что генотип CYP2E1*1С/ CYP2E1*1С ассоциирован с шейным дебютом БАС, тогда как наличие аллеля CYP2E1*1D достоверно коррелировало с более злокачественными формами БДН
— грудным и диффузным дебютами БАС и ПБП (χ2=3,85; ð=0,0492).
П о л и м о р ф и з м г е н а C Y P 2 D 6 . Частоты аллеля
CYP2D6 и его генотипов у больных с БДН и в контрольной группе приведены в табл. 3. Достоверных различий в частотах между российской и британской европеоидной контрольными группами выявлено не было. Например, частота гомозиготных носителей аллеля CYP2D6*4 составила 4%, как и в исследовании М. Brown и соавт. [12]. Частоты генотипов в основной и контрольной группах соответствовали уравнению Харди — Вейнберга.
Несмотря на ранее показанную ассоциацию между развитием БАС и наличием аллеля CYP2D6*4 [55], прямое сравнение с помощью критерия χ2 частот и генотипов аллеля CYP2D6 не выявило достоверных различий между группами больных с БДН и контролем в российской популяции (см. табл. 3). Однако количество гомозиготных носителей аллеля CYP2D6*4 в группе больных с БДН было повышено.
Поскольку гомозиготность по аллелю CYP2D6*4 приводит к фенотипу ММ [53], мы объединили гомозигот без аллеля CYP2D6*4 (а) и гетерозигот по аллелю CYP2D6*4 (б) в один генотип. Сравнительный анализ распределения комбинированного генотипа CYP2D6 (а+б) и гомозигот по аллелю CYP2D6*4 выявил достоверные различия между больными с БДН и контрольной группой (см. табл. 3). Гомозиготность
по аллелю CYP2D6*4 достоверно чаще отмечалась в группе БДН (р=0,04).
При изучении распределения комбинированного генотипа CYP2D6 у больных с разными клиническими характеристиками, как и при анализе аллеля CYP2E1, достоверных корреляций генотипов CYP2D6*4 полиморфизма с этими показателями обнаружено не было.
Полиморфизм генов GSTT1 и GSTM1. Частоты генотипов GSTT1 и GSTM1 в группе БДН и в контроле представлены в табл. 4. Необходимо отметить, что метод изучения полиморфизмов данных генов позволял убедительно судить лишь о наличии «нулевых» генотипов GSTT1(0/0) и GSTM1(0/0). Гетеро- и гомозиготные носители нормальных аллелей были объединены и проанализированы как генотипы GSTT1(+) или GТМ1(+).
Наблюдавшиеся частоты гомозиготных делеций GSTT1 и GSTM1 в контрольной популяции согласовывались с результатами других исследований в популяции европеоидов [8, 50, 57]. Доля носителей «нулевого» генотипа GSTT1 была значительно ниже частоты генотипа GSTM1(0/0) и сопоставима в обеих группах (см. табл. 4). Результаты анализа частоты генотипа GSTT1(0/0) отличались от таковых в исследовании М. Stroombergen и R. Waring [59]. В группе БДН отмечалось преобладание носителей «нулевого» генотипа GSTT1, однако в британской популяции ассоциации между гомозиготной делецией гена GSTM1 и развитием БДН не было установлено. В отличие от этого мы в российской популяции обнаружили достоверные различия в распределении генотипа GSTM1(0/0) между больными с БДН и контрольной группой. У больных частота генотипа GSTM1(0/0) была достоверно снижена, а генотипа GSTM1(+) — достоверно повышена (см. табл. 4). Более того, при анализе распределения комбинированных генотипов GSTT1/ GSTM1 обнаружены статистически значимые разли- чия между группами БДН и контроля (р=0,033) ввиду преобладания комбинаций генотипов GSTT1(0/0)/ GSTM1/(0/0) и GSTT1(+)/GSTM1(0/0) в группе контроля и комбинаций генотипов GSTT1(0/0)/ GSTM1(+) и GSTT1(+)/GSTM1(+) в группе БДН.
Таблица 3. Распределение аллелей и генотипов полиморфизма гена CYP2D6*4 у больных с БДН и в контрольной группе
Показатель |
|
ÁÄÍ |
|
Kонтроль |
χ2 |
p |
|
|
|
|
|
||||
n |
% |
n |
% |
||||
|
|
|
|||||
|
|
|
|
|
|
|
|
Частота аллелей |
|
|
|
|
|
|
|
He-CYP2D6*4 |
114 |
76,0 |
174 |
82,8 |
|
|
|
CYP2D6*4 |
36 |
24,0 |
36 |
17,2 |
2,57 |
0,11 |
|
Частота генотипов |
|
|
|
|
|
|
|
Non-CYP2D6*4/Non-CYP2D6*4 (a) |
49 |
65,3 |
74 |
70,5 |
|
|
|
Non-CYP2D6*4/CYP2D6*4 (á) |
16 |
21,3 |
26 |
24,8 |
|
|
|
CYP2D6*4/CYP2D6*4 |
10 |
13,3 |
5 |
4,7 |
4,28^ |
0,12^ |
|
à+á |
65 |
86,7 |
100 |
95,3 |
|
|
|
CYP2D6*4/CYP2D6*4 |
10 |
13,3 |
5 |
4,7 |
4,21 |
0,04 |
|
Всего |
75 |
— |
105 |
— |
|
|
|
|
|
|
|
||||
8 |
|
|
ЖУРНАЛ НЕВРОЛОГИИ И ПСИХИАТРИИ, 1, 2006 |
||||
Nv_1_06_.p65 |
8 |
20.12.2005, 10:38 |
ГЕНЕТИКА БОЛЕЗНИ ДВИГАТЕЛЬНОГО НЕЙРОНА
Таблица 4. Распределение генотипов делеционного полиморфизма генов GSTT1 и GSTМ1 у больных с БДН и в контрольной группе
Показатель |
ÁÄÍ |
|
Kонтроль |
χ2 |
p |
||
|
|
|
|
|
|||
|
n |
% |
n |
% |
|||
|
|
|
|
||||
|
|
|
|
|
|
|
|
Частота генотипов |
|
|
|
|
|
|
|
GSTT1(+) |
56 |
74,7 |
85 |
81,0 |
|
|
|
GSTT1(0/0) |
19 |
25,3 |
20 |
19,0 |
0,68 |
0,41 |
|
GSTM1(+) |
45 |
60,0 |
46 |
43,8 |
|
|
|
GSTM1(0/0) |
30 |
40,0 |
59 |
56,2 |
4,59 |
0,032 |
|
Частота комбинаций генотипов |
|
|
|
|
|
|
|
GSTT1(0/0)/GSTÌ1(0/0) |
8 |
10,7 |
16 |
15,2 |
|
|
|
GSTT1(+)/GSTÌ1(0/0) |
22 |
29,3 |
43 |
41 |
|
|
|
GSTT1(0/0)/GSTÌ1(+) |
11 |
14,7 |
4 |
3,8 |
|
|
|
GSTT1(+)/GSTÌ1(+) |
34 |
45,3 |
42 |
40 |
8,75^ |
0,033^ |
|
Всего |
75 |
— |
105 |
— |
|
|
|
|
|
|
|
|
|
|
|
Таблица 5. Частота аллелей |
и распределение |
генотипов |
Ile105Val-полиморфизма гена |
GSTP1 ó |
больных с БДН |
||
и в контрольной группе |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Показатель |
ÁÄÍ |
|
Kонтроль |
χ2 |
p |
||
|
|
|
|
|
|||
|
n |
% |
n |
% |
|||
|
|
|
|
||||
|
|
|
|
|
|
|
|
Частота аллелей |
|
|
|
|
|
|
|
GSTP1*A |
99 |
66 |
146 |
69,5 |
|
|
|
GSTP1*B |
51 |
34 |
64 |
30,5 |
0,50 |
0,48 |
|
Частота генотипов |
|
|
|
|
|
|
|
GSTP1*A/GSTP1*B |
31 |
41,3 |
48 |
45,7 |
|
|
|
GSTP1*A/GSTP1*B (a) |
37 |
49,3 |
50 |
47,6 |
|
|
|
GSTP1*B/GSTP1*B (á) |
7 |
9,3 |
7 |
6,7 |
0,60^ |
0,74^ |
|
GSTP1*A/GSTP1*A |
31 |
41,3 |
48 |
45,7 |
|
|
|
à+á |
44 |
58,7 |
57 |
54,3 |
0,34 |
0,56 |
|
Всего |
75 |
— |
105 |
— |
|
|
|
|
|
|
|
|
|||
Примечание. ^ — χ2 рассчитан при помощи программы R½C. |
|
|
|
|
|||
Достоверных корреляций между наличием «нуле- |
дованиях было показано, что ферменты, содержащие |
вых» генотипов GSTT1 и GSTM1 и клиническими ха- |
валин в 105-м кодоне, характеризуются нарушением |
рактеристиками БДН выявлено не было. |
термостабильности и менее активно катализируют |
Полиморфизм гена GSTP1. Результаты генотипи- |
некоторые ксенобиотики [24, 65], мы объединили |
рования гена GSTP1 представлены в табл. 5. Анализ 13 |
гетерозиготных (а) и гомозиготных (б) носителей |
новых больных не внес изменений в ранее опублико- |
аллеля GSTP1*В в одну генотипическую категорию с |
ванные нами данные [1]. Чаще всего в группах БДН и |
наличием валина в 105-м кодоне. Однако, как видно |
контроля встречался аллель дикого типа (GSTP1*A). |
из табл. 5, распределение комбинированных геноти- |
Наблюдаемая частота гомозиготности по аллелю |
пов GSTP1 (а+б) и гомозигот по аллелю GSTP1*А |
GSTP1*В в контрольной группе была сопоставима с |
было сходным. |
ранее опубликованными результатами генотипирова- |
Достоверных корреляций между наличием ком- |
ния в популяции европеоидов [25, 52]. Распределение |
бинированного генотипа GSTP1 и клиническими ха- |
генотипов GSTP1 соответствовало уравнению Харди |
рактеристиками БДН, приведенными в табл. 1, не |
— Вейнберга. Определялось некоторое преобладание |
установлено. |
гомозиготных носителей аллеля GSTP*B в группе |
Обнаружена лишь ассоциация между вариантами |
БДИ, однако оно не являлось статистически досто- |
поражения мотонейронов и наличием комбинирован- |
верным (см. табл. 5). Поскольку в предыдущих иссле- |
ного генотипа GSTP1 (γ-корреляция: r=2,909, p=0,003; |
ЖУРНАЛ НЕВРОЛОГИИ И ПСИХИАТРИИ, 1, 2006 |
9 |
Nv_1_06_.p65 |
9 |
20.12.2005, 10:38 |
ОБЩИЕ ВОПРОСЫ НЕВРОЛОГИИ И ПСИХИАТРИИ
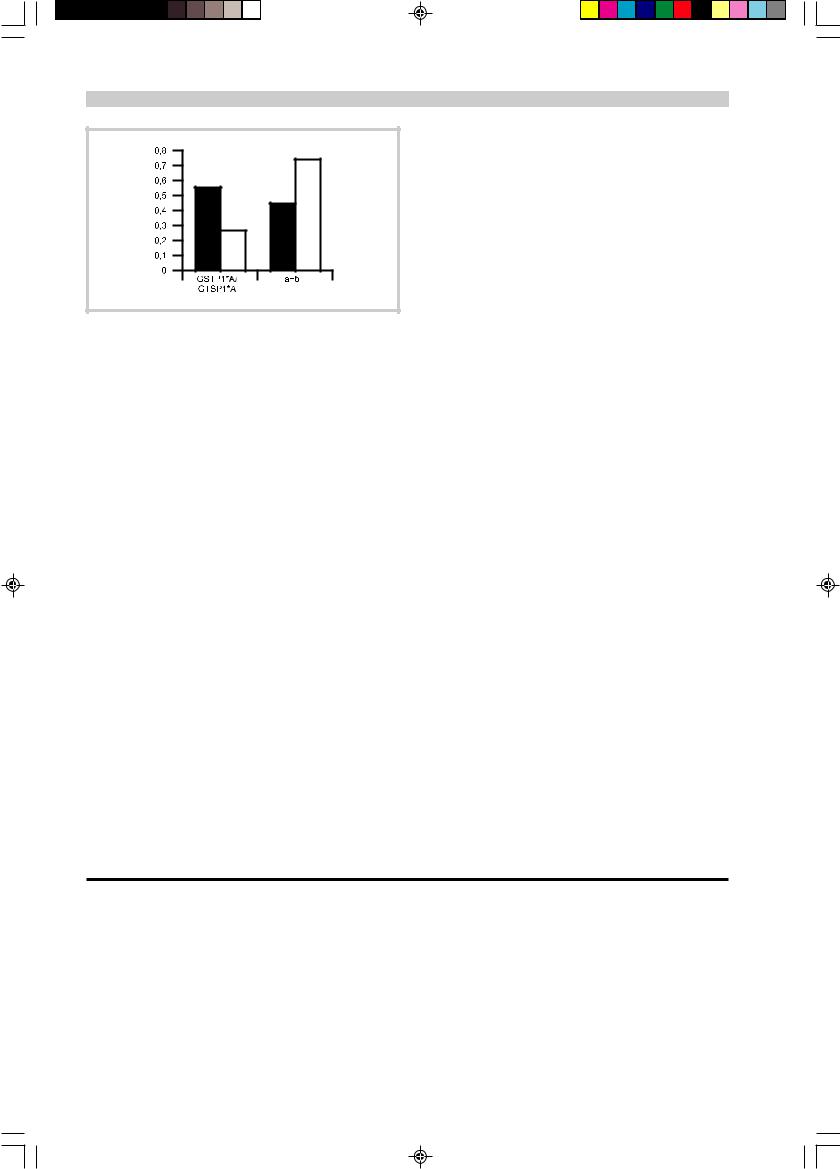
Распределение генотипов Ile105Val-полиморфизма гена GSTP1 у больных с разными вариантами БДН.
Темные столбцы — классический вариант, светлые — сегментарноядерный и пирамидный варианты БДН.
корреляция Спирмена: r=0,242, p=0,036). Дальнейший анализ показал, что наличие аллеля дикого типа (GSTP1*A/GSTP1*A) ассоциировано с классическим вариантом БДН (равномерным поражением центральных и периферических мотонейронов), тогда как наличие аллеля GSTP1*В достоверно коррелировало с сегментарно-ядерным и пирамидным вариантами БДН, при которых страдают преимущественно либо периферические, либо центральные мотонейроны соответственно (χ2=6,60; р=0,0102; см. рисунок).
Полиморфизм гeнa NAT2. У больных с БДН и в контрольной группе проводили генотипирование NAT2. Выявили наиболее распространенные «медленные» аллели S1, S2 и S3 (NAT2*5, NAT2*6 и NAT2*7 соответственно) и «быстрый» аллель дикого типа F1 (NAT2*4). Частоты аллелей NAT2 и генотипов NAT2, которые соответствовали уравнению Харди — Вейнберга, приведены в табл. 6.
Количество аллелей S1, S2 и S3 и частоты различных генотипов, проанализированные в отдельности, были достаточно низкими (данные не приведены), поэтому мы объединили аллели в две группы («медленные» — S и «быстрые» — F1) и все генотипы разделили на два комбинированных генотипа (быстрые ацетиляторы и медленные и промежуточные ацетиляторы). Распределение быстрых ацетиляторов, как и медленных и промежуточных, в контрольной группе не отличалось от ранее установленного в популяции европеоидов [15]. Частота медленных и промежуточных ацетиляторов была несколько выше в
группе больных БДН, однако прямое сравнение с применением критерия χ2 не выявило достоверных различий частот комбинаций аллелей и генотипов NAT2 в группах больных и контроля (см. табл. 6). Достоверных корреляций между типами ацетиляторов и клиническими характеристиками БДН не обнаружено.
Обсуждение
Цель настоящего исследования заключалась в оценке возможного участия полиморфизмов отдельных генов системы детоксикации и их комбинаций в патогенезе БДН. Молекулярно-генетический анализ семейной формы БДН показал, что в патогенезе заболевания участвует несколько генов. Однако мутации в этих генах объясняют лишь 10% случаев семейной и спорадической БДН. В большинстве случаев БДН ее первичные причины не установлены. Это обусловливает необходимость выявления других генов, участвующих в патогенезе БДН. Эти гены могут запускать развитие нейродегенерации или же служить факторами предрасположенности к ее развитию в со- четании с другими генетическими факторами или факторами окружающей среды, такими как действие ксенобиотиков. Одним из значимых патогенетических факторов БДН являются свободные радикалы [35]. Это дает основание полагать, что в патогенезе БДН могут участвовать процессы детоксикации. Гены, кодирующие ферменты, которые метаболизируют ксенобиотики, характеризуются значительным полиморфизмом, и наличие делеций или «медленных» аллелей может приводить к дисбалансу процессов детоксикации. CYP, GSTs и NAT2 играют важную роль в развитии многих заболеваний и метаболизме ксенобиотиков.
Важную роль в защите организма от действия ксенобиотиков играют CYP [21]. Ранее было показано, что CYP2E1 и CYP2D6 могут участвовать в патогенезе неврологических заболеваний [11, 17, 51, 56]. CYP2E1 (этанолиндуцибельный фермент) характеризуется инсерционным полиморфизмом 96 п.о. (CYP2E1*1D), наличие которого ассоциировано с более высокой индуцированной активностью по сравнению с диким типом [38]. Ассоциация между полиморфизмом CYP2E1*1D и развитием БДН ранее не изучалась. В настоящем исследовании показано, что частота аллеля CYP2E1*1D значительно повышена у больных БДН (14% при 2,5% в контроле; р<0,001). Обнаруже-
Таблица 6. Частота генотипов NAT2 у больных с БДН и в контрольной группе
Частота ацетилятора |
|
ÁÄÍ |
|
Kонтроль |
χ2 |
|
|
|
|
|
|
p |
|||
n |
% |
n |
% |
||||
|
|
|
|||||
|
|
|
|
|
|
|
|
Аллели F1 |
35 |
23,3 |
56 |
26,7 |
|
|
|
Аллели S |
115 |
76,7 |
154 |
73,3 |
1,49 |
0,47 |
|
Быстрые ацетиляторы |
32 |
42,7 |
52 |
49,5 |
|
|
|
Медленные и промежуточные |
43 |
57,3 |
53 |
50,5 |
0,83 |
0,36 |
|
ацетиляторы |
|
|
|
|
|
|
|
Всего |
75 |
— |
105 |
— |
|
|
|
|
|
|
|
||||
10 |
|
|
ЖУРНАЛ НЕВРОЛОГИИ И ПСИХИАТРИИ, 1, 2006 |
||||
Nv_1_06_.p65 |
10 |
20.12.2005, 10:38 |
на также достоверно более высокая частота гомозиготных носителей аллеля CYP2E1*1D и комбинированного генотипа CYP2E1 (CYP2E1*1D гетерозиготы
èCYP2E1*1D гомозиготы) среди больных с БДН в российской популяции. Эти данные говорят о нали- чии ассоциации между полиморфизмом CYP2E1*1D
èразвитием спорадической БДН, а также об участии аллеля CYP2E1*1D в патогенезе спорадической БДН в российской популяции больных. CYP2E1 — это белок, продуцирующий свободные радикалы [66]. Он обладает способностью индуцировать катализируемую ионом железа реакцию Фентона и усиливать опосредованный гидроксил-радикалами метаболизм некоторых ксенобиотиков, особенно этанола [16]. Индукция этого фермента в астроцитах приводила к развитию оксидантного стресса, увеличивала продукцию метаболитов перекисного окисления липидов и снижала концентрацию глутатиона [41]. Эти процессы могут участвовать в дегенерации мотонейронов. Мы также установили, что наличие аллеля CYP2E1*1D достоверно коррелирует с развитием наиболее злока- чественных форм БДН, таких как грудной и диффузный дебюты БАС и ПБП. Показано, что при одной и той же мутации гена Cu-, Zn-зависимой супероксиддисмутазы (СОД-1) в рамках одной семьи смогут развиваться разные фенотипы БДН [20]. Продукты модифицирующих генов или факторов предрасположенности могут отчасти способствовать такой фенотипической вариабельности. Этот феномен может объяснять выявленные нами корреляции между вариантами генотипа CYP2E1*1D и клиническими характеристиками БДН.
Другим цитохромом, который может участвовать в патогенезе БДН, является CYP2D6. Снижение его активности наследуется по аутосомно-рецессивному типу, и носителей данного аллеля относят к группе ММ — «медленной метаболизации» [29]. Аллель CYP2D6*4 ассоциирован со снижением активности CYP2D6 [53]. Анализ этого полиморфизма показал, что гомозиготные носители аллеля CYP2D6*4 достоверно чаще встречаются среди больных с БДН. В другом исследовании ассоциаций с развитием БДН ус-
тановлено достоверное преобладание частоты аллеля CYP2D6*4 при БДН [56]. Замена G→A на границе 3-го интрона и 4-го экзона гена, которая и лежит в основе данного аллельного варианта [22], нарушает сплайсинг РНК, что в свою очередь приводит к преждевременному завершению трансляции и к образованию дефектного продукта, лишенного ферментативной активности. Указанный патологический продукт данного гена может обладать новыми свойствами, прямо или косвенно способствующими дегенерации мотонейронов. Это говорит о том, что наличие аллеля CYP2D6*4 может быть фактором риска развития БДН в российской популяции.
Метаболиты фазы I детоксикации часто токсич- нее первичных соединений, и важно, чтобы они не накапливались. Ферменты фазы II, такие как GST и NAT, преобразуют соединения, синтезированные в фазу I в нетоксичные продукты [48]. Ферменты GST предотвращают развитие оксидантного стресса, инактивируя токсичные соединения и свободные радикалы посредством конъюгации с глутатионом. Делеци-
ЖУРНАЛ НЕВРОЛОГИИ И ПСИХИАТРИИ, 1, 2006
ГЕНЕТИКА БОЛЕЗНИ ДВИГАТЕЛЬНОГО НЕЙРОНА
онные варианты генов GSTT1 и GSTM1, а также Ilе105Val-полиморфизм гена GSTP1 в его 5-м экзоне служат предрасполагающими факторами к развитию различных заболеваний, в частности хронического бронхита, атеросклероза, разных видов рака [26] и неврологических заболеваний [59]. Поскольку токсины окружающей среды и оксидантный стресс принимают участие в патогенезе неврологических заболеваний, снижение протективных свойств глутатион- S-трансфераз при наличии «нулевых» вариантов изоформ GSTT1 и GSTM1, а также валинсодержащей изоформы GSTP1 может быть ассоциировано с развитием БДН. Анализ распределения генотипов в контрольной группе российской популяции соответствует ранее опубликованным зарубежным данным. Анализ Ile105Val-полиморфизма гена GSTP1 не обнаружил различия в распределении генотипов GSTP1 между группами БДН и контроля. Однако нами установлено, что генотип, содержащий аллель дикого типа (изолейцин в 105-м кодоне), ассоциирован с класси- ческим вариантом БДН (равномерным поражением центральных и периферических мотонейронов), тогда как наличие в этом кодоне валина ассоциировано с наличием сегментарно-ядерного и пирамидного вариантов заболевания. Ферменты, кодируемые аллелями 105Val, характеризуются термолабильностью и снижением специфической активности [24, 65], а больные с аллелем 105Val страдают менее злокачественными вариантами поражения мотонейронов. Поэтому возможно, что ген GSTP1 способен оказывать модифицирующее влияние на фенотип БДН в российской популяции.
Проведенный нами анализ распределения генотипов GSTT1 и GSTM1 показал, что частота генотипа GSTM1(0/0) достоверно снижена, а генотипа GSTM1(+) достоверно повышена у больных с БДН. Помимо делеционного аллеля, существуют еще два распространенных аллеля — GSTM1*А и GSTM1*В, в структуре которых имеются замены пар оснований. Предполагается, что гомо- и гетерозиготы по аллелям GSTM1*А и GSTM1*В подвержены различным заболеваниям в меньшей степени по сравнению с носителями генотипа GSTM1(0/0) [19, 23], а гетерозиготный генотип АВ может оказывать протективное влияние в отношении БДН по аналогии с протективным эффектом в отношении рака прямой кишки и опухолей головного мозга, который характерен для данных генотипов [19]. Достоверное снижение частоты генотипа GSTM1(0/0) у обследованных нами больных может быть вызвано сцеплением аллеля GSTM1(0) с протективными аллелями GSTM1*А и GSTM1*В или гетерозиготными генотипами. Методика ПЦР, примененная в настоящем исследовании, не позволяла различить аллели GSTM1*А и GSTM1*В, однако использование другого метода на большем количестве больных в будущем может помочь в решении этой проблемы.
Другим важным участником фазы II является ариламин-N-ацетилтрансфераза 2-то типа (NAT2), которая катализирует N-ацетилирование большого числа ксенобиотиков. Многие работы свидетельствуют о влиянии полиморфизма ацетилирования NAT2 на развитие различных заболеваний, в том числе некото-
11
Nv_1_06_.p65 |
11 |
20.12.2005, 10:38 |
ОБЩИЕ ВОПРОСЫ НЕВРОЛОГИИ И ПСИХИАТРИИ
рых видов рака [4, 28, 34] и неврологических, например болезни Паркинсона [7]. В одном из исследований [43] cообщается о повышении частоты полиморфизма «медленных ацетиляторов» в группе БДН по сравнению с контрольной группой в Великобритании, однако эта закономерность не была подтверждена после введения поправки на множественные сравнения. Полученные нами результаты подтверждают эти данные. Мы установили, что в частоте комбинаций аллелей и генотипов NAT2 не имеется достоверных различий между группами БДН и контроля. Это позволяет предположить, что полиморфизм ацетилирования NAT2 не участвует в патогенезе БДН у обследованных нами больных.
В заключение считаем возможным высказать предположение, что в развитии спорадической БДН у больных из Российской Федерации участвуют по крайней мере 4 гена I и II фаз детоксикации — СYР2E1,
CYP2D6, GSTP1 è GSTM1.
Исследование отчасти поддержано грантом Российского фонда фундаментальных исследований (гранты ¹ 04-04-49510, 04-04-08117), программой Российской Академии Наук «Физико-химическая биология», грантом Президента Российской Федерации в поддержку молодых ученых России (грант МК-3047.2004.4) и грантом Президента Российской Федерации в поддержку ведущих научных школ (грант ScS-1430.2003.4).
ЛИТЕРАТУРА
1.Жеребцова А.Л., Шадрина М.И., Левицкий Г.Н. и др. Анализ Ilel05Val полиморфизма гена глутатион-S-трансферазы при спорадиче- ской болезни двигательного нейрона в Российской Федерации. Генетика 2004; 6: 69.
2.Cкворцова В.И., Лимборская С.А., Сломинский П.А. и др. Особенности спорадической болезни двигательного нейрона, ассоциированной с мутациями D90A и G12R, в российской популяции. Журн неврол и психиат 2003; 103: 46—52.
3.Abdel-Rahman S.Z.,el-Zein R.A., Anwar W.A.,Au W.W.A multiplex PCR procedure for polymorphic analysis of GSTM1 and GSTT1 genes in population studies. Cancer Lett 1996; 107: 229—233.
4.Ambrosone C.B., Freudencheim J.L., Graham S. et al. Cigarette smoking, N-acetyltransferase 2 genetic polymorphisms, and breast cancer risk. J Am Med Assoc 1996; 276: 1494—1512.
5.Andersen P.M. Genetics of sporadic ALS. Amyotroph Lateral Scler Other Motor Neuron Disord 2001; 2: 37—41.
6.Andersen P.M., Sims K.B., Xin W.W. et al. Sixteen novel mutations in the Cu/Zn superoxide dismutase gene in amyotrophic lateral sclerosis: a decade of discoveries, defects and disputes. Amyotroph Lateral Scler Other Motor Neuron Disord 2003; 4: 62—73.
7.Bandmann O., Vaughan J.R., Holmans P. et al. Detailed genotyping demonstrates association between the slow acetylator genotype for N-acetyltransferase 2 (NAT2) and familial Parkinson’s disease. Mov Disord 2000; 15: 30—35.
8.Baranova H., Canis M., Ivaschenko T. et al. Possible involvement of arylamine N-acetyltransferase 2, glutathione S-transferases Ml and Tl genes in the development of endometriosis. Mol Hum Reprod 1999; 5: 636—641.
9.Bertz R.J., Granneman G.R. Use of in vitro and in vivo data to estimate the likelihood of metabolic pharmacokinetic interactions. Clin Pharmacokinet 1997; 32: 210—258.
10.Bikmaeva A.R., Sibiriak S.V., Khusnutdinova E.K. Insertional polymorphism of the CYP2E1 gene in infiltrative pulmonary tuberculosis in populations of Bashkortostan Republic. Mol Biol 2004; 38: 239—243.
11.Borroni Â., Archetti S., Agosti C. et al. Intronic CYP46 polymorphism along with ApoE genotype in sporadic Alzheimer Disease: from risk factors to disease modulators. Neurobiol Aging 2004; 25: 747—751.
12.Brown M.A., Edwards S., Hoyle E. et al. Polymorphisms of the CYP2D6 gene increase susceptibility to ankylosing spondylitis. Hum Mol Genet 2000; 9: 1563—1566.
13.Cacabelos R. The application of functional genomics to Alzheimer’s disease. Pharmacogenomics 2003; 4: 597—621.
14.Cascorbi I., Drakoulis N., Brockmoller J. et al. Arylamine N-acetyl- transferase (NAT2) mutations and their allelic linkage in unrelated Caucasian individuals: correlation with phenotypic activity. Am J Hum Genet 1995; 57: 581—592.
16.Dupont I., Lucas D., Clot P. et al. Cytochrome P4502E1 inducibility and hydroxyethyl radical formation among alcoholics. J Hepatol 1998;
28:564—571.
17.Forsyth J.T., Grünewald R.A., Rostami-Hodjegan A. et al. Parkinson’s disease and CYP1A2 activity. Br J Clin Pharmacol 2000; 50: 303— 309.
18.Fritsche E., Pittman G., Bell D.A. Localization, sequence analysis, and ethnic distribution of a 96-br insertion in the promoter of the human CYP2E1 gene. Mutation Research Genomics 2000; 432: 1—5.
19.Fryer A.A., Zhao L., Alldersea J. et al. Use of site-directed mutagenesis of allele-specific PCR primers to identify the GSTM1A, GSTM1B, GSTM1 A,B and GSTM1 null polymorphisms at the glutathione S-transferase, GSTM1 locus. Biochem J 1993; 295: 313— 315.
20.Giess R., Holtmann B., Braga M. et al. Early onset of severe familial amyotrophic lateral sclerosis with a SOD-1 mutation: potential impact of CNTF as a candidate modifier gene. Am J Hum Genet 2002;
70:1277—1286.
21.Gonzalez F.J. The molecular biology of cytochrome P450s. Pharmacol Rev 1988; 40: 243—288.
22.Gough A.C., Miles J.S., Spurr N.K. et al. Identification of the primary gene defect at the cytochrome P450 CYP2D locus. Nature 1990;
347:773—776.
23.Harada S., Misawa S., Nakamura T.„et al. Detection of GSTI gene deletion by the polymerase chain reaction and its possible correlation with stomach cancer in Japanese. Hum Genet 1992; 90: 62—64.
24.Harries L.W., Stubbins M.J., Forman D. et al. Identification of genetic polymorphisms at the glutathione S-transferase Pi locus and association with susceptibility to bladder, testicular and prostate cancer. Carcinogenesis 1997; 18: 641—644.
25.Harris M.J., Coggan M., Langton L. et al. Polymorphism of the Pi class glutathione S-transferase in normal populations and cancer patients. Pharmacogenetics 1998; 8: 27—31.
26.Hatagima A. Genetic polymorphisms and metabolism of endocrine disruptors in cancer susceptibility. Cad Saude Publica 2002; 18: 357— 377.
27.Hein D.W., Doll M.A., Rustan T.D. et al. Metabolic activation and deactivation of arylamine carcinogens by recombinant human NAT1 and polymorphic NAT2 acetyltransferases. Carcinogenesis 1993; 14: 675—678.
28.Hung R.J., Boffetta P., Brennan P. et al. GST, NAT, SULT1A1, CYP1B1 genetic polymorphisms, interactions with environmental exposures and bladder cancer risk in a high-risk population. Int J Cancer 2004; 10: 598—604.
29.Ingelman-Sundberg M., Oscarson M., McLellan R.A. Polymorphic human cytochrome P450 enzymes: an opportunity for individualized drug treatment. Trends Pharmacol Sci 1999; 20: 342—349.
15.Daly A.K., Cholerton S., Armstrong M., Idle J.R. Genotyping for poly30. James C.M., Daniels J., Wiles C.M., Owen M.J. Debrisoquine hydrox-
morphisms in xenobiotic metabolism as a predictor of disease susceptibility. Environ Health Perspect 1994; 102: 55—61.
ylase gene polymorphism in motor neuron disease. Neurodegeneration 1994; 3: 149—152.
12 |
ЖУРНАЛ НЕВРОЛОГИИ И ПСИХИАТРИИ, 1, 2006 |
Nv_1_06_.p65 |
12 |
20.12.2005, 10:38 |
31.Landi S. Mammalian class theta GST and differential susceptibility to carcinogens: a review. Mutat Res 2000; 463: 247—283.
32.Leigh P.N., Ray-Chaudhuri K. Motor neuron disease. J Neurol Neurosurg Psychiat 1994; 57: 886—896.
33.Lieber C.S. Cytochrome P4502E1: its physiological and pathological role. Physiol Rev 1997; 77: 517—544.
34.Lincz L.F., Kerridge I., Scorgie F.E. et al. Xenobiotic gene polymorphisms and susceptibility to multiple myeloma.Haematologica2004;
89:628—629.
35.Liu D., Wen J., Liu J., Li L. The roles of free radicals in amyotrophic lateral sclerosis: reactive oxygen species and elevated oxidation of protein, DNA, and membrane phospholipids. FASEB J 1999; 13: 2318—2328.
36.Majoor-Krakaauer D., Willems P.J., Hofman A. Genetic epidemiology of amyotrophic lateral sclerosis. Clin Genet 2003; 63: 83—101.
37.Mannervik Â. The isoenzymes of glutathione transferase. In: Advances in Enzymology. Ed. A. Meister. New York: Wiley and Sons 1985;
57:357—417.
38.McCarver D.J., Byun R., Hines R.N. et al. A genetic polymorphism in the regulatory sequences of human CYP2E1: association with increased chlorzoxazone hydroxylation in the presence of obesity and ethanol intake. Toxicol Appl Pharmacol 1998; 152: 276—281.
39.McCarver D.G. ADH2 and CYP2E1 genetic polymorphisms: risk factors for alcohol-related birth defects. Drug Metab Dispos 2001; 29: 562—565.
40.Miller S.A., Dykes D.D., Polesky H.F. A sample salting out procedure for extracting DNA from human nucleated cells. Nucleic Acid Res 1988; 16: 1215.
41.Montoliu Ñ., Sancho-Tello M., Azorin I. et al. Ethanol increases cytochrome P4502E1 and induces oxidative stress in astrocytes. J Neurochem 1995; 65: 2561—2570.
42.Nelson L.M. Epidemiology of ALS. Clin Neurosci 1996; 3: 327—331.
43.Nicholl D.J., Bennett P., Hiller L. et al. A study of five candidate genes in Parkinson’s disease and related neurodegenerative disorders. European Study Group on Atypical Parkinsonism. Neurology 1999;
53:1415—1421.
44.Noor R., Mittal S., Iqbal J. Supperoxide dismutase — applications and relevance to human diseases. Med Sci Monit 2002; 8: 210—215.
45.Norris F.H., Callachini P.R., Fallat R.G. et al. Administration of guanidine in amyotrophic lateral sclerosis. Neurology 1974; 24: 721— 728.
46.Norris F., Shepherd R., Denys E. et al. Onset, natural history and outcome in idiopathic adult motor neuron disease. J Neurol Sci 1993; 118: 48—55.
47.Pickett C.B., Lu A.Y. Glutathione S-transferases: gene structure, regulation, and biological function. Annu Rev Biochem 1989; 58: 743— 764.
48.Raunio H., Husgafvel-Pursiainen K., Anttila S. et al. Diagnosis of polymorphisms in carcinogen-activating and inactivating enzymes and cancer susceptibility. Gene 1995; 159: 113—121.
49.Raymond M.L., Rousset F. An exact test for population differentiation. Evolution 1995; 49: 1280—1283.
ЖУРНАЛ НЕВРОЛОГИИ И ПСИХИАТРИИ, 1, 2006
ГЕНЕТИКА БОЛЕЗНИ ДВИГАТЕЛЬНОГО НЕЙРОНА
50.Rebbeck T.R. Molecular epidemiology of the human glutathione S- transferase genotypes GSTM1 and GSTT1 in cancer susceptibility. Cancer Epidemiol Biomarkers Prev 1997; 6: 733—743.
51.Riedl A.G., Watts P.M., Jenner P. et al. P450 enzymes and Parkinson’s disease: the story so far. Mov Disord 1998; 13: 212—220.
52.Saarikoski S.T., Voho A., Reinikainen M. et al. Combined effect of polymorphic GST genes on individual susceptibility to lung cancer. Int J Cancer 1998; 77: 516—521.
53.Sachse Ñ., Brockmoller J., Bauer S., Roots I. Cytochrome P450 2D6 variants in a Caucasian population: allele frequencies and phenotypic consequences. Am J Hum Genet 1997; 60: 284—295.
54.Santt O., Baranova H., Albuisson E. et al. Interaction between GSTMlnull and CYP2D6-deficient alleles in the pathogenesis of Parkinson’s disease. Eur J Neurol 2004; 11: 247—251.
55.Seidegard J., Vorachek W.R., Pero R.W., Pearson W.R. Hereditary differences in the expression of the human glutathione transferase active on trans-stilbene oxide are due to a gene deletion. Proc Natl Acad Sci USA 1988; 85: 7293—7297.
56.Siddons M.A., Pickering-Brown S.M., Mann D.M.„et al. Debrisoquine hydroxylase gene polymorphism frequencies in patients with amyotrophic lateral sclerosis. Neurosci Lett 1996; 208: 65—68.
57.Spielberg S.P. N-acetyltransferases: pharmacogenetics and clinical consequences of polymorphic drug metabolism. J Pharmacokinet Biopharm 1996; 24: 509—519.
58.Steventon G.B., Heafield M.T., Waring R.H., Williams AC. Xenobiotic metabolism in Parkinson’s disease. Neurology 1989; 39: 883—887.
59.Stroombergen M.C., Waring R.H. Determination of glutathione S- transferase mu and theta polymorphisms in neurological disease. Hum Exp Toxicol 1999; 18: 141—145.
60.Spurr N.K., Gough A.C., Chinegwundoh F.I., Smith C.A. Polymorphisms in drug-metabolizing enzymes as modifiers of cancer risk. Clin Chem 1995; 41: 1864—1869.
61.Suzuki Ò., Coggan M., Shaw D.V., Board P.G. Electrophoretic and immunological analysis of human glutathione S-transferase isozymes. Annals of Human Genetics 1987; 51: 95—106.
62.Waring R.H., Steventon G.B., Sturman S.G. et al. S-methylation in motorneuron disease and Parkinson’s disease. Lancet 1989; 2: 356— 357.
63.Watson M.A., Stewart R.K., Smith G.B. et al. Human glutathione S- transferase P1 polymorphisms: relationship to lung tissue enzyme activity and population frequency distribution. Carcinogenesis 1998; 19: 275—280.
64.Wolf C.R., Dale Smith C.A., Gough A.C. et al. Relationship between the debrisoquine hydroxylase polymorphism and cancer susceptibility. Carcinogenesis 1992; 13: 1035—1038.
65.Zima T., Fialova L., Mestek O. et al. Oxidative stress, metabolism of ethanol and alcohol-related diseases. J Biomed Sci 2001; 8: 59—70.
66.Zimniak P., Nanduri Â., Pikula S. et al. Naturally occurring human glutathione S-transferase GSTP1-1 isoforms with isoleucine and valine in position 104 differ in enzymic properties. Eur J Biochem 1994; 224: 893—899.
Поступила 22.09.05
13
Nv_1_06_.p65 |
13 |
20.12.2005, 10:38 |