

Журнал неврологии и психиатрии / 2006 / NEV_2006_01_02
.pdfКЛИНИКА НЕРВНЫХ И ПСИХИЧЕСКИХ ЗАБОЛЕВАНИЙ
Болезни нижнего двигательного нейрона с преимущественным поражением верхних конечностей: это самостоятельные формы или атипичные варианты бокового амиотрофического склероза?
Т.Р. СТУЧЕВСКАЯ, В.М. КАЗАКОВ, Д.И. РУДЕНКО, О.В. ПОСОХИНА, А.А. СКОРОМЕЦ
Lower motor neuron diseases with predominantly upper limbs affection: are they independent forms or atypical variants of amyotrophic lateral sclerosis?
T.R. STUCHEVSKAYA, V.M. KAZAKOV, D.I. RUDENKO, O.V. POSOKHINA, A.A. SKOROMETS
ГМПБ ¹2, Санкт-Петербург; Санкт-Петербургский государственный медицинский университет им. И.П. Павлова
Проведен анализ 5 спорадических случаев болезни нижнего двигательного нейрона с преимущественным поражением проксимальных отделов рук у двух больных и кистей у трех. Представленные наблюдения, а также описанные в литературе аналогичные случаи с преимущественным поражением рук, разными течением заболевания и денервационными изменениями при проведении игольчатой ЭМГ могут рассматриваться как проявления клинической гетерогенности болезни нижнего двигательного нейрона. На ранних этапах заболевания определяются сложности дифференциальной диагностики атипичных вариантов бокового амиотрофического склероза — БАС (например, синдромом «свисающих рук») и первичной прогрессирующей спинальной мышечной атрофии взрослых. Можно предположить, что атипичные варианты БАС, в основе которых лежит поражение только нижнего мотонейрона (синдром «свисающих рук» и атрофия одной кисти), являются самостоятельной нозологической единицей, отличной от БАС.
Ключевые слова: болезни двигательного нейрона с преимущественным поражением верхних конечностей, боковой амиотрофический склероз.
We carried out an analysis of 5 sporadic cases of lower motor neuron disease with predominant affection of the proximal parts of arms in 2 patients and distal parts in 3 patients. From clinical point of view, our own observations, along with similar cases reported in the literature with predominantly affected upper limbs, different progression of the disease and denervation changes during needle EMG, can argue for clinical heterogeneity of lower motor neuron disease. There were some difficulties in establishment of a differential diagnosis between atypical variants of amyotrophic lateral sclerosis (“flail arm” syndrome) and primary muscular atrophy of adults at the early stages of the disease. We suppose that atypical variants of amyotrophic lateral sclerosis resultant from affection of lower motor neuron only (“flail arm” syndrome and distal amyotrophy), could be distinguished from amyotrophic lateral sclerosis and considered as an independent entity.
Key words: lower motor neuron diseases with predominantly upper limbs affection, amyotrophic lateral sclerosis.
В последние годы в классификацию болезней нижнего мотонейрона были включены различные формы с преимущественным вовлечением верхних конечностей [11]. Самостоятельность некоторых из них подтверждена данными молекулярно-генетических исследований (например, спинальная мышечная атрофия с преимущественным поражением дистальных отделов верхних конечностей, тип V) [1]. Однако нозологическая принадлежность ряда других болезней нижнего мотонейрона, таких как брахиальная
© Коллектив авторов, 2006
Zh Nevrol Psikhiatr Im SS Korsakova 2006;106: 1: 14—20
диплегия, или синдром свисающих рук, болезнь Хирояма, или атрофия одной конечности, остается не до конца ясной: являются ли они отдельными нозологическими формами болезней нижнего мотонейрона наследственной или экзогенной природы либо всего лишь атипичные варианты бокового амиотрофического склероза (БАС)?
Мы наблюдали 5 больных с атрофией и слабостью верхних конечностей на момент начала болезни с характерными признаками вовлечения нижнего мотонейрона (см. таблицу). Разная локализация мышечных поражений в начале заболевания, особенности распространения мышечных атрофий и слабости по мере его развития и быстроте прогрессирования процесса позволили предположить, что наши больные по клиническим признакам составляют гетеро-
14 |
ЖУРНАЛ НЕВРОЛОГИИ И ПСИХИАТРИИ, 1, 2006 |
Nv_1_06_.p65 |
14 |
20.12.2005, 10:38 |
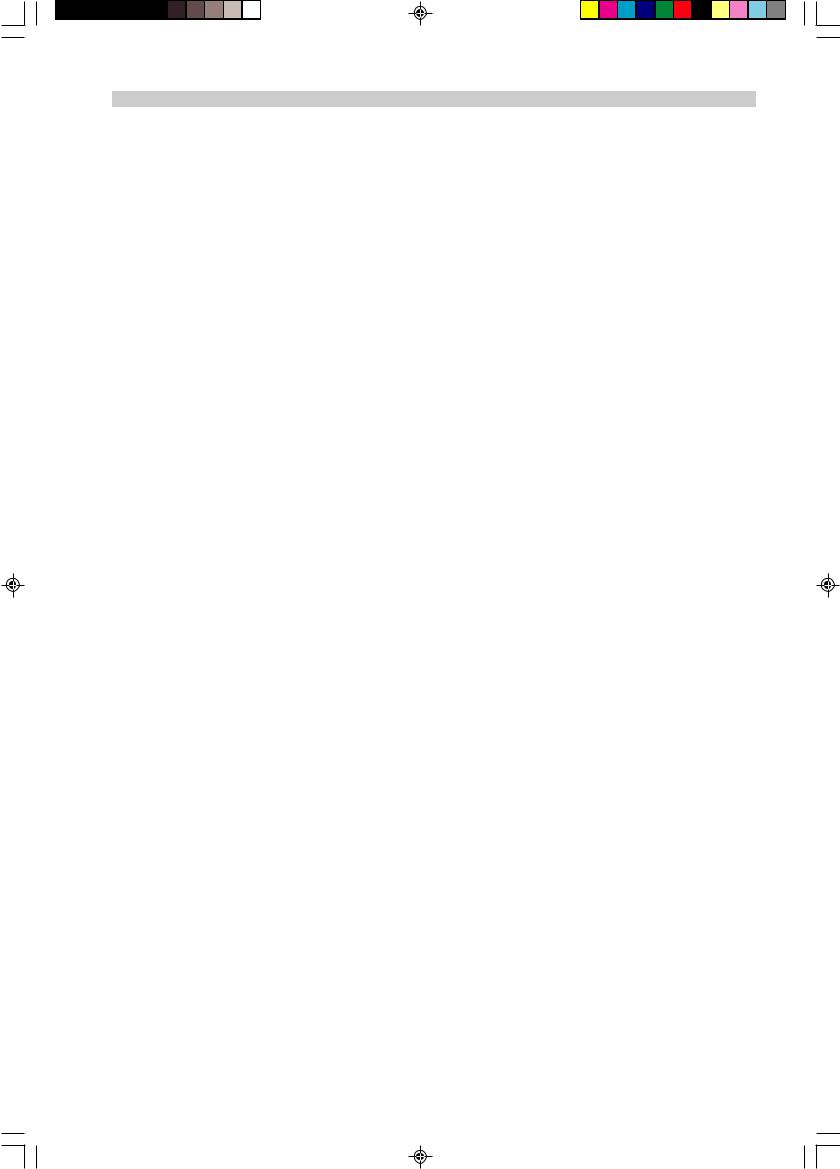
генную группу пациентов с болезнями нижнего мотонейрона, возможно, разными по этиологии и патогенезу.
Материал и методы
Больных разделили на две группы. I группу составили двое мужчин в возрасте 48 (пациент 1) и 83 (пациент 2) лет с симметричной слабостью проксимальных мышц рук. Во II группу включили двух мужчин в возрасте 16 (пациент 3) и 44 (пациент 4) лет и женщину 33 лет (пациент 5) с асимметричной слабостью мышц кистей. Течение заболевания было быстро прогрессирующим у пациента 1, медленно прогрессирующим у пациента 2 и стабильным (стационарным) у пациентов 3—5.
Для диагностики использовали игольчатую электромиографию (ЭМГ), измерение скорости проведения импульса по двигательным и чувствительным волокнам периферических нервов на верхних и нижних конечностях, МРТ шейного отдела позвоночника, генеалогический анамнез и определение уровня креатинфосфокиназы (КФК) в сыворотке крови.
Результаты и обсуждение
Больные I группы
Как видно из таблицы, особенностями мышеч- ного паттерна у больных этой группы было раннее, тяжелое и симметричное поражение надплечий и проксимальных отделов рук: надостных, подостных, дельтовидных, двуглавых и трехглавых мышц (руки «свисали» в положении пронации предплечий и кистей; движения в плечевых суставах почти отсутствовали, а в локтевых были резко ограничены). В последующем наблюдалось постепенное распространение слабости и атрофий на мышцы предплечий и кистей. Межлопаточные мышцы были относительно сохранны: больные могли привести лопатки к позвоночнику и лопатки не отступали от грудной клетки. Глубокие рефлексы на руках отсутствовали, на ногах были высокими. Фасцикуляции на руках отмечались только у пациента 1.
Через год от начала болезни пациенты I группы не могли работать с поднятыми руками, принимать пищу самостоятельно из-за отсутствия движений в руках и нуждались в постоянной посторонней помощи.
Через 2,5 года от начала болезни у пациента 1 отмечена генерализация процесса: наряду с вялой верхней параплегией выявлялась выраженная слабость мышц живота, тазового пояса, задней группы мышц бедер с сохранением силы в четырехглавых мышцах. Он не мог вставать с кровати, кресла, подниматься по лестнице без посторонней помощи. Коленные рефлексы снизились, ахилловы оставались высокими. В дальнейшем на протяжении 3 лет мышечная слабость нарастала, больной перестал ходить, появились расстройства дыхания и больной умер от остановки дыхания в возрасте 53 лет.
У пациента 2 через 2,5 года не наблюдалось распространения мышечной слабости за пределы надплечий и рук. Он мог свободно спускаться и подниматься по лестнице, совершать прогулки по улице.
ЖУРНАЛ НЕВРОЛОГИИ И ПСИХИАТРИИ, 1, 2006
БОЛЕЗНЬ ДВИГАТЕЛЬНОГО НЕЙРОНА
Бульбарных симптомов, патологических кистевых
èстопных знаков, нарушения чувствительности, координации, а также функции тазовых органов у больных I группы выявлено не было.
Клинический и биохимический анализы крови и мочи были без отклонений от нормы. Уровень КФК в крови у обоих пациентов был в пределах нормы. Антитела GM1, GD1b в крови у пациента 1 обнаружены не были, у пациента 2 не исследовались.
Гормоны щитовидной железы (Т3 св, T4 св, ТТГ) были в пределах нормы значений. Патологии эндокринной системы, включая гинекомастию, ни у первого, ни у второго больного не отмечалось.
Исследование цереброспинальной жидкости: без отклонений от нормы.
Наследственный анамнез у обоих больных без особенностей.
Электрофизиологическое исследование. При иголь- чатой ЭМГ у обоих больных выявлялись нейрогенные изменения в мышцах рук и ног. При ЭМГ круговой мышцы рта и языка отклонений от нормы не выявлялось. Кондакционного блока обнаружено не было (см. таблицу).
МРТ шейного отдела позвоночника. У обоих пациентов отмечались дегенеративно-дистрофические изменения в шейном отделе позвоночника, осложненные грыжеобразованием без компрессии спинного мозга и корешков. Изменения интенсивности сигнала от спинного мозга выявлено не было (см. таблицу).
Клинические данные и результаты ЭМГ (денервационные изменения на руках) давали повод предположить, что в основе болезни лежит нейрогенный процесс. Отсутствие отчетливых клинических признаков поражения пирамидного тракта на момент нача- ла заболевания, спинномозговых корешков, периферических нервов, собственно мышц и данные ЭМГ позволили связать имеющиеся двигательные нарушения с поражением передних рогов спинного мозга. Снижение скорости проведения импульса по чувствительным волокнам периферических нервов у пациента 2 можно объяснить субклинической сенсорной полиневропатией пожилого возраста.
Â2001 г. мы предположили наличие у обоих больных прогрессирующей спинальной мышечной атрофии взрослых с преимущественным поражением рук. Однако у пациента 1 заболевание быстро прогрессировало и через год после появления первых симптомов развилась двусторонняя брахиальная диплегия с последующим поражением мышц тазового пояса и бедер. Учитывая быстрый темп течения болезни, мы не могли полностью исключить у этого больного наличие атипичного варианта БАС, представленного как прогрессирующая мышечная атрофия с преимущественным поражением рук, напоминающего синдром «свисающих рук» (flail arm syndrome).
У пациента 2 до сих пор сохраняется фенотип синдрома «свисающих рук», т.е. мышечная слабость
èатрофии не распространяются на ноги и/или бульбарные мышцы. В настоящее время у этого больного нельзя полностью исключить наличия спинальной мышечной атрофии взрослых с преимущественным поражением верхних конечностeй (рис. 1).
15
Nv_1_06_.p65 |
15 |
20.12.2005, 10:38 |

КЛИНИКА НЕРВНЫХ И ПСИХИЧЕСКИХ ЗАБОЛЕВАНИЙ
Клинические и инструментальные данные 5 пациентов
|
I группа |
|
II группа |
|
||
Параметр |
|
|
|
|
|
|
пациент 1 |
пациент 2 |
пациент 3 |
пациент 4 |
пациент 5 |
||
|
||||||
|
|
|
|
|
|
|
Ïîë |
Ì |
Ì |
Ì |
Ì |
Æ |
|
Возраст |
52 |
85 |
27 |
48 |
35 |
|
Возраст начала |
48 |
83 |
16 |
44 |
33 |
|
заболевания, годы |
|
|
|
|
|
|
Течение заболевания |
5 ëåò (óìåð) |
Прогрессирует |
Не прогрессирует |
Не прогрессирует 2 года |
Не прогрессирует |
|
|
|
|
более 6 лет |
|
1 ãîä |
|
Мышечный паттерн |
Проксималь- |
Проксимальные |
Kисть правая, |
Kисть левая |
Kисть правая |
|
на начало |
ные отделы рук |
отделы рук |
дистальная часть |
|
|
|
заболевания (первого |
и надплечья |
и надплечья |
правого |
|
|
|
осмотра) |
|
|
предплечья |
|
|
|
Симметрично/ |
Асимметрично |
Симметрично |
Форма monomelic |
Форма monomelic |
Форма monomelic |
|
асимметрично |
(в начале |
|
|
|
|
|
|
болезни) |
|
|
|
|
|
SCM |
Сохранны |
Сохранны |
Сохранны |
Сохранны |
Сохранны |
|
Бульбарные |
Íåò |
Íåò |
Íåò |
Íåò |
Íåò |
|
симптомы на момент |
|
|
|
|
|
|
начала болезни |
|
|
|
|
|
|
и в последующем |
|
|
|
|
|
|
Распределение |
Проксимально |
Проксимально |
Дистально |
Дистально |
Дистально |
|
мышечной слабости |
|
|
|
|
|
|
и атрофий |
|
|
|
|
|
|
Фасцикуляции |
На руках |
Íå áûëî |
Íå áûëî |
Íå áûëî |
Íå áûëî |
|
Рефлексы |
Отсутствовали |
Отсутствовали на |
Снижены |
Высокие на руках и ногах (D>S) |
Высокие на руках и |
|
|
на руках, были |
руках и ахилловы, |
|
|
ногах |
|
|
повышены |
коленные |
|
|
|
|
|
на ногах |
повышены |
|
|
|
|
Патологические |
Íåò |
Íåò |
Íåò |
Íåò |
Россолимо — |
|
знаки |
|
|
|
|
Вендеровича с двух |
|
|
|
|
|
|
сторон, |
|
|
|
|
|
|
непостоянные знаки |
|
|
|
|
|
|
Бабинского |
|
Чувствительность |
Не нарушена |
Не нарушена |
Не нарушена |
Гипалгезия по ульнарной части |
Не нарушена |
|
|
|
|
|
левой кисти |
|
|
ÝÌÃ |
Нейрогенные |
Нейрогенные |
Нейрогенные |
Нейрогенные изменения в |
Нейрогенные |
|
|
изменения в |
изменения в |
изменения в |
межкостных мышцах левой кисти |
изменения в мышцах |
|
|
мышцах рук и |
мышцах рук и ног. |
мышцах правой |
и в 1-м межкостном промежутке |
правой кисти, |
|
|
íîã |
Снижение |
кисти, |
правой кисти, снижена скорость |
предплечья. |
|
|
|
скорости |
предплечья |
проведения импульса по |
При симуляционных |
|
|
|
проведения по |
|
чувствительным волокнам |
пробах снижение |
|
|
|
чувствительным |
|
локтевого нерва (слева до 32 м/с, |
амплитуды М-ответов |
|
|
|
волокнам |
|
справа до 35 м/с), и двигательным |
при стимуляции |
|
|
|
периферических |
|
волокнам локтевых нервов на |
двигательных |
|
|
|
нервов на руках и |
|
предплечье (слева до 33 м/с, |
волокон n. ulnaris |
|
|
|
ногах: |
|
справа до 40 м/с) cо снижением |
и n. medianus справа |
|
|
|
n. medianus 33 ì/ñ, |
|
амплитуд М-ответа по |
|
|
|
|
n. ulnaris 34 ì/ñ, |
|
чувствительным волокнам |
|
|
|
|
n. suralis 30 ì/ñ |
|
периферических нервов (слева |
|
|
|
|
|
|
до 2,4 мкВ, справа до 2,4 мкВ), |
|
|
|
|
|
|
по двигательным волокнам |
|
|
|
|
|
|
периферических нервов (слева |
|
|
|
|
|
|
до 1,1 мВ, справа до 5,3 мВ) |
|
|
|
|
|
|
|
|
|
|
Продолжение таблицы на след. стр. |
16 |
ЖУРНАЛ НЕВРОЛОГИИ И ПСИХИАТРИИ, 1, 2006 |
Nv_1_06_.p65 |
16 |
20.12.2005, 10:38 |
БОЛЕЗНЬ ДВИГАТЕЛЬНОГО НЕЙРОНА
Продолжение таблицы
|
I группа |
|
II группа |
|
||
Параметр |
|
|
|
|
|
|
пациент 1 |
пациент 2 |
пациент 3 |
пациент 4 |
пациент 5 |
||
|
||||||
|
|
|
|
|
|
|
МРТ шейного отдела |
Грыжи |
Остеофиты на |
Левосторонняя |
Задние грыжи межпозвонковых |
Без отклонений от |
|
позвоночника |
межпозвонко- |
уровне С5, артрит |
парамедианная |
дисков С2 — С7 без компрессии |
нормы |
|
|
вых дисков С4 |
фасеточных |
грыжа диска |
корешков и спинного мозга. |
|
|
|
— Ñ5, Ñ5/Ñ6 è |
суставов; |
С3 — С4. Задние |
Сигнал от спинного мозга не |
|
|
|
Ñ6/Ñ7 áåç |
гипертрофия |
протрузии дисков |
изменен |
|
|
|
компрессии |
желтой связки, |
Ñ4 — Ñ5 è |
|
|
|
|
спинного мозга |
гиперлордоз на |
Ñ5 —Ñ6. |
|
|
|
|
и корешков; |
уровне С3; |
Медиальная |
|
|
|
|
сигнал от |
сужение переднего |
грыжа С6—С7 |
|
|
|
|
спинного мозга |
субарахноидально- |
без компрессии |
|
|
|
|
не изменен |
го пространства |
корешков. |
|
|
|
|
|
на уровне С4 — С7 |
Уменьшение |
|
|
|
|
|
без компрессии |
переднезаднего |
|
|
|
|
|
спинного мозга. |
диаметра |
|
|
|
|
|
Сигнал от |
спинного мозга |
|
|
|
|
|
спинного мозга |
справа на уровне |
|
|
|
|
|
не изменен |
Ñ6 — Ñ7 |
|
|
|
Антитела GM1 |
Не выявлены |
Не исследовались |
Не исследовались |
Не исследовались |
Не исследовались |
|
|
|
|
|
|
|
|
à |
á |
Рис. 1. Пациент 2. Спинальная мышечная атрофия взрослых с поражением верхних конечностей.
а — симметричная тяжелая атрофия и слабость надостных, подостных, дельтовидных, двуглавых и трехглавых мышц; б — руки «свисают» в положении пронации предплечий (фенотип синдрома «свисающих рук»).
Следует отметить, что на ранних этапах болезни достаточно трудно дифференцировать синдром «свисающих рук» — атипичный вариант БАС без признаков поражения пирамидного тракта в нижних конеч- ностях и прогрессирующую спинальную мышечную атрофию взрослых. Однако быстрое прогрессирование болезни и высокие рефлексы на ногах у пациента 1 позволяют исключить первичную прогрессирующую спинальную мышечную атрофию взрослых. Между тем описаны случаи прогрессирующей спинальной мышечной атрофии взрослых с вовлечением пирамидного тракта; с другой стороны, у больного с синдромом «свисающих рук» (вариант БАС) в тече- ние ряда лет процесс может не распространяться на
бульбарные мышцы, мышцы тазового пояса и ног и могут отсутствовать признаки вовлечения пирамидного тракта [4, 16].
Таким образом, создается впечатление, что в первые месяцы заболевания нет отчетливых клинических признаков, которые позволяли бы отличить синдром «свисающих рук» (атипичный вариант БАС) от прогрессирующей спинальной мышечной атрофии взрослых.
Имеются сложности в проведении дифференциальной диагностики вариантов БАС — «синдрома свисающих рук» и прогрессирующей мышечной атрофии (предполагаемый БАС по критериям El. Escorial). Например, у пациента 1 в ранних стадиях болезни (ког-
ЖУРНАЛ НЕВРОЛОГИИ И ПСИХИАТРИИ, 1, 2006 |
17 |
Nv_1_06_.p65 |
17 |
20.12.2005, 10:38 |
КЛИНИКА НЕРВНЫХ И ПСИХИЧЕСКИХ ЗАБОЛЕВАНИЙ
да развилась двусторонняя брахиальная диплегия) можно было поставить диагноз БАС, фенотип синдрома «свисающих рук». Однако в более поздних стадиях заболевания клиническая картина у этого больного соответствовала прогрессирующей мышечной атрофии с преимущественным поражением верхних конечностей, слабостью мышц туловища и ног без пирамидных знаков. Таким образом, у пациента 1 начальный фенотип «свисающих рук» трансформировался в прогрессирующую мышечную атрофию.
Из литературы известно, что при аутопсии больных, умерших вследствие прогрессирующей мышеч- ной атрофии, находили дегенерацию нейронов моторной коры, включая клетки Беца, кортико-спи- нального тракта и уменьшение количества клеток передних рогов спинного мозга и двигательных ядер ствола мозга, что подтверждает диагноз достоверного БАС, несмотря на клинический диагноз, предполагаемый БАС (по критериям El. Escorial) [2, 16]. Таким образом, получается так, что некоторые случаи болезни моторного нейрона могут быть представлены ограниченным распределением мышечной слабости и атрофий в надплечьях и проксимальных отделах рук без признаков поражения пирамидного тракта с разным в последующем течением заболевания (например, присоединение бульбарных симптомов или распространение процесса на ноги и туловище). По-видимому, пациенты с таким ограниченным мышечным паттерном в начале болезни без признаков поражения верхнего мотонейрона с последующей быстрой генерализацией процесса должны рассматриваться как имеющие атипичный вариант БАС.
Однако полностью нельзя исключить того, что больные с фенотипом прогрессирующей мышечной атрофии и длительным течением заболевания (более 7—10 лет) без присоединения признаков поражения верхнего двигательного нейрона страдают первичной прогрессирующей спинальной мышечной атрофией взрослых. Ответить на эти вопросы, возможно, помогут молеку- лярно-генетические исследования больных с атипич- ными вариантами болезни двигательного нейрона.
Практически у всех наблюдавшихся нами пациентов с болезнью двигательного нейрона имелись де- генеративно-дистрофические изменения шейного отдела позвоночника, осложненные грыжеобразованием. Подобные поражения шейного отдела позвоноч- ника могут служить непосредственной причиной атрофий и слабости мышц плечевого пояса и проксимальных отделов рук или могут только усиливать симптомы и признаки болезни двигательного нейрона. Однако последующее развитие атрофий и слабости в мышцах ног у некоторых таких пациентов, отсутствие сенсорных расстройств и наличие ЭМГ-призна- ков парциальной денервации как в пораженных, так в непораженных конечностях, а также отсутствие признаков компрессии спинного мозга при МРТ могут свидетельствовать о наличии у них болезни двигательного нейрона, а не спондилогенной шейной миелопатии.
Больные II группы
У пациентов 3, 4 и 5 клиническая картина была представлена тяжелой атрофией и слабостью мелких
мышц правой или левой кисти. Только у пациента 3 отмечалось распространение атрофий на дистальный отдел правого предплечья. Глубокие рефлексы на руках были снижены у пациента 3 и патологически высоки у пациентов 4 и 5. Фасцикуляций, бульбарных симптомов, признаков поражения респираторных мышц, патологических стопных знаков, нарушений чувствительности, координации и функции тазовых органов выявлено не было. Болезнь развилась у пациента 3 во второй, у пациента 4 в третьей, у пациентки 5 в четвертой декаде жизни. Атрофия и слабость прогрессировали в течение 6 лет у пациента 3, в течение 3 лет у пациентов 4 и 5, после чего произошла стабилизация симптомов и признаков заболевания.
Клинический и биохимический анализы крови и мочи у больных II группы были без отклонений от нормы. Уровень КФК в крови во всех случаях был в пределах нормы.
Исследование цереброспинальной жидкости: без отклонений от нормы.
Наследственный анамнез у всех пациентов без особенностей.
Патологии эндокринной системы ни у одного из больных не отмечалось.
Электрофизиологическое исследование. При иголь- чатой ЭМГ у пациентов 3 и 5 выявлялись нейрогенные изменения только в мышцах правой пораженной кисти, у пациента 4 — в правой и левой кисти. При проведении стимуляционных проб у пациента 4 была снижена скорость проведения импульса по чувствительным и двигательным волокнам локтевых нервов со снижением амплитуд М-ответов. При ЭМГ круговой мышцы рта и языка отклонений от нормы не выявлено. Кондакционного блока обнаружено не было (см. таблицу).
МРТ шейного отдела позвоночника. У всех пациентов отмечались дегенеративно-дистрофические изменения в шейном отделе позвоночника, осложненные грыжеобразованием без компрессии спинного мозга и корешков; лишь у пациента 3 наблюдалось уменьшение переднезаднего диаметра спинного мозга справа на уровне С6 — С7. Изменения интенсивности сигнала от спинного мозга не было выявлено.
Итак, клинические данные и результаты ЭМГ (денервационные изменения в пораженных мышцах) позволили диагностировать в начале заболевания у пациентов II группы сегментарную дистальную мышечную атрофию, в основе которой может лежать как нейрональный, так и невральный процесс. Так, у пациента 4 при электронейромиографии отмечалось снижение амплитуд М-ответов и скорости проведения импульсов по чувствительным и двигательным волокнам локтевого нерва на предплечье слева и справа. Однако паттерн мышечной слабости выходил за рамки иннервации только локтевого нерва, а также не было клинических признаков поражения правой руки. В течение последних 2 лет заболевание не прогрессирует. Больному был установлен диагноз мультифокальной моторной невропатии без кондакционного блока. Дифференциальная диагностика проводилась с болезнью Хираяма, атипичным вариантом БАС, прогрессирующей спинальной мышечной атрофией взрослых (дистальный тип).
18 |
ЖУРНАЛ НЕВРОЛОГИИ И ПСИХИАТРИИ, 1, 2006 |
Nv_1_06_.p65 |
18 |
20.12.2005, 10:38 |

Пациентка 5 с тяжелой атрофией мышц правой кисти, патологически высокими рефлексами на руках и ногах, непостоянными двусторонними знаками Бабинского, быстро прогрессирующим течением в начале заболевания и денервационными изменениями на ЭМГ, что клинически соответствовало monomelic БАС (БАС с поражением одной конечности). Однако в течение последнего года болезнь не прогрессирует. В связи с этим нельзя исключить наличия у этой больной прогрессирующей спинальной мышеч- ной атрофии взрослых (дистальный тип), ассоциированной с поражением пирамидного тракта, фенотипически напоминающей БАС (рис. 2).
У пациента 3 слабость и атрофии появились в мелких мышцах правой кисти и позднее распространились на дистальную часть предплечья пораженной руки. Слабость и атрофии прогрессировали в течение 6 лет, после чего наступила полная стабилизация симптомов и признаков заболевания. Последние 7 лет болезнь не прогрессирует. С учетом клиники и данных ЭМГ этому больному был поставлен диагноз фокальной амиотрофии дистальной части верхней конечности, или болезни Хираяма.
Справка из истории
Термин «непрогрессирующая ювенильная фокальная амиотрофия дистальной части верхней конечности» был впервые предложен K. Hirayama в 1959 г. [5]. Болезнь характеризуется односторонней (редко двусторонней) прогрессирующей мышечной слабостью и неврогенными атрофиями мышц кисти и предпле- чья. Прогрессирование болезни продолжается в тече- ние нескольких лет, после чего происходит стабилизация признаков и симптомов.
Развитие мышечных атрофий и слабости связывают с поражением передних рогов спинного мозга, средних и нижних шейных сегментов [7]. Однако патогенез поражения спинного мозга до сих пор неясен. В литературе обсуждается несколько возможных механизмов поражения шейного отдела спинного мозга. Одной из главных причин заболевания на сегодняшний день считается хроническая микроциркуляторная недостаточность. Однако причины этой патологии пока недостаточно изучены.
В 1987 г. К. Hirayama и соавт. при проведении аутопсии пациента с ювенильной фокальной амиотрофией (умершего от других причин) выявили признаки ишемического некроза (уменьшение нейронов с глиозом). Обнаруженные морфологические изменения позволили авторам высказать предположение об ишемической миелопатии на уровне от С5 до Th1 сегментов спинного мозга как основном факторе, приводящем к развитию заболевания [6].
При последующих исследованиях шейного отдела позвоночника с помощью КТ, МРТ и миелографии у больных с фокальной амиотрофией верхних конечностей в некоторых случаях выявлялась только умеренная атрофия в нижнешейном отделе спинного мозга, но никогда не сообщалось об интрамедуллярных изменениях спинного мозга, характерных для ишемической миелопатии [9, 12].
Ряд авторов [17] при проведении МРТ шейного отдела позвоночника в положении сгибания шеи у
ЖУРНАЛ НЕВРОЛОГИИ И ПСИХИАТРИИ, 1, 2006
БОЛЕЗНЬ ДВИГАТЕЛЬНОГО НЕЙРОНА
à
á
â
Рис. 2. Пациентка 5. Фокальная амиотрофия кисти (болезнь Хираяма).
Тяжелая атрофия и слабость мышц тыльных и ладонных межкостных, возвышения I и V пальцев правой кисти.
некоторых больных с болезнью Хираяма и в контрольной группе выявляли умеренное уменьшение переднезаднего диаметра спинного мозга в нижнешейных сегментах. У ряда пациентов отмечались выраженные
19
Nv_1_06_.p65 |
19 |
20.12.2005, 10:38 |

КЛИНИКА НЕРВНЫХ И ПСИХИЧЕСКИХ ЗАБОЛЕВАНИЙ
дегенеративно-дистрофические изменения в шейном отделе позвоночника (протрузии межпозвонковых дисков, ретроспондилолистез). У части больных эти изменения приводили к отчетливой компрессии спинного мозга во время сгибания шеи, но не было отме- чено ассоциации с ипсилатеральными клиническими расстройствами (слабостью и атрофиями) или изменениями в спинном мозге. Эти наблюдения позволили авторам предположить, что ювенильная фокальная амиотрофия верхних конечностей является собственно дегенеративным заболеванием моторных нейронов. Ранее такая точка зрения была высказана U. Misra и Kalita [10], которые исследовали показатели центральной скорости проведения по кортико-мышеч- ному пути с помощью транскраниальной магнитной стимуляции и не выявили признаков, характерных для ишемической миелопатии.
К. Hirayama и Y. Tokumaru [8], проведя серию функциональных нейрорадиологических исследований у 73 пациентов с ювенильной фокальной амиотрофией в возрасте от 11 до 19 лет, обнаружили признаки динамической компрессии нижнешейного отдела спинного мозга во время сгибания шеи. На основании полученных данных авторы предположили, что ювенильная фокальная амиотрофия верхних конечностей является особым типом шейной миелопатии, которая развивается вследствие смещения дурального мешка во время сгибания шеи и компрессионного уплощения нижнешейного отдела спинного мозга. Такое же мнение о патогенезе заболевания было высказано другими исследователями, которые выявили характерное для компрессионной миелопатии уменьшение амплитуды пика N13 только у больных с болезнью Хираяма при исследовании соматосенсорных вызванных потенциалов с рук в нейтральной позиции и при максимальном сгибании шеи [13].
Соотношение болезни Хираяма с группой прогрессирующих спинальных мышечных атрофий взрослых, ювенильной формой БАС и его атипичным ва-
риантом (monomelic БAC) также остается не вполне ясным. Нельзя исключить, что болезнь Хираяма является вариантом БАС. Аргументом в пользу этого может быть описание бельгийской семьи с наследственной формой БАС, ассоциированной с мутацией в гене SOD1 [14]. У одного из членов этой семьи была непрогрессирующая амиотрофия дистальной части одной руки. Этот факт позволил неврологам предположить, что болезнь Хираяма может быть связана с наследственным БАС, ассоциированным с мутацией
âгене супероксиддисмутазы (SOD1). Позднее были описаны два случая наследственной болезни Хираяма, но без мутации в гене SOD1 [15]. Однако, по мнению авторов, это не исключает гипотезы, что болезнь Хираяма является аллельным вариантом БАС.
Наряду с этим появилось сообщение о 49-летнем мужчине, у которого в возрасте 45 лет развилась прогрессирующая амиотрофия дистальной части левой верхней конечности без нарушений чувствительности [3]. У этого пациента c 35 лет отмечалась асимметричная нейросенсорная тугоухость, как у его матери и старшей сестры. В биоптатах мышц были обнаружены ЦОК-негативные волокна, однако шероховатых красных волокон выявлено не было. У этого пациента была выявлена мутация в митохондриальной ДНК, в гене 7472insC, кодирующем tRNASer (UCN). Это второе сообщение, документирующее ассоциацию заболевания нижнего моторного нейрона (фенотип фокальной амиотрофии, болезни Хираяма) со специфиче- ской мутацией в митохондриальной ДНК.
Таким образом, мы представили 5 клинически гетерогенных случаев заболевания нижнего мотонейрона, которые, возможно, обусловлены генетическими или гормональными нарушениями. Дальнейшие клинические, иммунологические и генетические исследования этих форм заболевания способны помочь
âразграничении пациентов с заболеванием нижнего двигательного нейрона, у которых никогда не разовьется БАС, и больных с БАС.
ЛИТЕРАТУРА
1.Auer-Grumbach M., Loscher W.N., Wagner K. et al. Phenotypic and genotypic heterogeneity in hereditary motor neuronopathy type V. Brain 2000; 8: 1612—1623.
2.Ince P.G., Evans J., Knopp M. et al. Corticospinal tract degeneration in the progressive muscular atrophy variant of ALS. Neurology 2003;
60:1252—1258.
3.Fetoni V., Briem E., Carrara F. et al. Monomelic amyotrophy associated with the 7472insC mutation in the mtDNA tRNASer (UCN) gene. Neuromuscular Disorders 2004; 14: 723—726.
4.Gent E.V., Hoogland R.A., Jennekens FGI. Distal amyotrophy of predominantly the upper limbs with pyramidal features in the large kinship.Neurology 1985; 48: 266—269.
5.Hyrayama K., Tsubaki T., Toyokura Y. et al. Juvenile muscular atrophy of unilateral upper extremity. Neurology 1963; 13: 373—380.
6.Hirayama K., Tomonaga M., Kitano K. et al. Focal cervical poliopathy causing juvenile muscular atrophy of distal upper extremity: a pathological study. J Neurol Neurosurg Psychiat 1987; 50: 285—290.
7.Hirayama K. Non-progressive juvenile spinal muscular atrophy of the distal upper limb. In: P.J. Vinken, G.W. Bruyn, H.L. Klawans (eds.). Handbook of Clinical Neurology. Amsterdam, the Netherlands: Elsevier Science Publishers 1991; 59: 107—120.
8.Hirayama K., Tokumaru Y. Cervical dural sac and spinal cord in juvenile muscular atrophy of distal upper extremity. J Neurol 2000;
54:1922—1926.
20
9.Karnaze M.G., Gado M.H., Sartor K.J. et al. Comparison of MR and CT myelography in imaging the cervical and thoracic spine. AJNR 1987; 8: 983—989.
10.Misra U.K., Kalita. Central motor conduction in Hirayama disease. Electroencephalogr Clin Neurophysiol 1995; 97: 73—76.
11.Neuromuscular Disease Center, Washington University School of Medicine, St.Louis, Mo. Hereditary motor syndromes (SMA, ALS+). 2003; http://www.neuro.wustl.edu/neuromuscular.
12.Norman D. et al. Magnetic resonance imaging if the central nervous system. New York: Raven Press 1987; 289—328.
13.Restuccia D., Rubino M., Valeriani M. et al. Cervical cord dysfunction during neck flexion in Hirayama’s disease. J Neurol 2003; 60: 1980—1983.
14.Robberecht W., Aguirre T., Van Den Bosch L. et al. D90A heterozygosity in the SOD1 gene is associated with familial and apparently sporadic amyotrophic lateral sclerosis. Neurology 1996; 47: 1336— 1339.
15.Robberecht W., Aguirre T., Bosch L. et al. Familial juvenile focal amyotrophy of theu upper extremity. Arch Neurol 1997; 54: 46—50.
16.Sasali S., Iwata M. Atypical form of amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiat 1999; 66: 581—585.
17.Schroder R., Keller E., Flacke S. et al. MRI findings in Hirayama disease: flexion-induced cervical myelopathy or intrinsic motor neuron disease? J Neurol 1999; 246: 1069—1074.
Поступила 29.04.05
ЖУРНАЛ НЕВРОЛОГИИ И ПСИХИАТРИИ, 1, 2006
Nv_1_06_.p65 |
20 |
20.12.2005, 10:38 |