

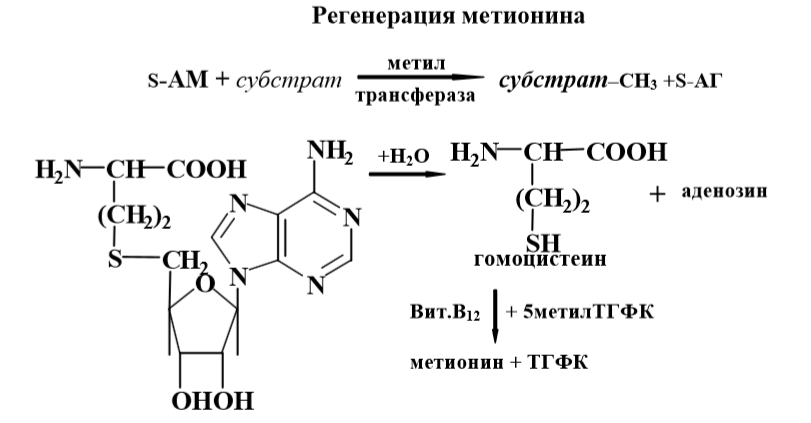
6.Трансметилирование. Метионин и s-аденозилметионин (участие в синтезе креатина, адреналина, фосфатидилхолина, метилировании чужеродных соединений). Участие тетрагидрофолиевой кислоты в метилировании.
Трансметилирование – это транспорт метильной группы от источника метильных групп к субстрату метилирования с помощью доноров или переносчиков метильных групп.
Источниками метильных групп служат серин, холин, бетаин. Переносчиками или донорами метильных групп для субстратов служат активная форма фолиевой кислоты – тетрагидрофолиевая кислота (ТГФК) и активная форма метионина – S-аденозилметионин
(S-АМ). Схема трансметилирования

Метильная группа переносится в 5-ом положении – 5-метил
ТГФК, но ТГФК может переносить и другие одноуглеродные фрагмен-
ты: N5 N10 метилен ТГФК (–СН 2–); N5 N10 метенил ТГФК (–СН=) и др.
В большинстве процессов биосинтеза донором метильной груп-
пы служит S-АМ.В результате S-АМ является универсальным донором метильных группдля самых разнообразных субстратов метилирования. Получает ме-тильную группу S-АМ от небольшого числа молекул – главным образом, от 5-метил ТГФК, реже – от холина и бетаина. Цикл активированной –СН3-группы

Примеры реакций метилирования
Синтез креатина идет в 2 стадии. В почках и поджелудочной железе образуется гуанидин-ацетат:

Креатин из печени разносится кровью к другим органам. В мышцах, особенно в сердце, он участвует в переносе энергии путем обратимого перефосфорилирования с АТФ с образованием макроэргического соединения – креатинфосфата (субстратное фосфорилирование). 2. Синтез адреналина – происходит в мозговом веществе надпо-
чечников в результате метилирования норадреналина.

Инактивация катехоламинов происходит двумя путями – дезаминированием с участием МАО и метилированием с участием катехол-О-метилтрансферазы. 3. Синтез фосфатидилхолина

Фосфатидилхолин необходим для построения мембран, в печени – для образования липопротеинов очень низкой плотности и липопротеинов высокой плотности. При дефиците фосфатидилхолина в печени накапливаются нейтральные жиры и развивается жировой гепатоз.
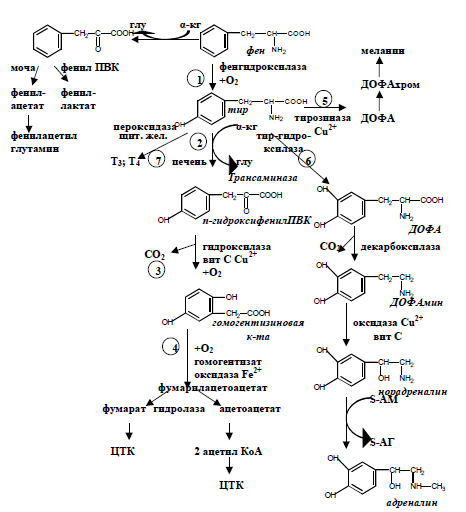
7. ОБМЕН ФЕНИЛАЛАНИНА И ТИРОЗИНА. ИСПОЛЬЗОВАНИЕ ТИРОЗИНА ДЛЯ СИНТЕЗА КАТЕХОЛАМИНОВ, ТИРЕОИДНЫХ ГОРМОНОВ, МЕЛАНИНА. РАСПАД ТИРОЗИНА ДО ФУМАРОВОЙ И АЦЕТОУКСУСНОЙ КИСЛОТ. НАСЛЕДСТВЕННЫЕ НАРУШЕНИЯ ОБМЕНА ФЕНИЛАЛАНИНА И ТИРОЗИНА.
Обмен фенилаланина и тирозина в норме
Фенилаланин – незаменимая аминокислота, у здоровых людей почти весь фенилаланин, который не был использован для синтеза белка, превращается в печени в тирозин.Очень незначительная часть фенилаланина трансаминируется с образованием фенилпиро-виноградной кислоты.
Тирозин – заменимая аминокислота, она используется для синте-за белка, для образования гормонов щитовидной железы, катехолами-нов, меланина, оставшийся тирозин подвергается трансаминированию и дальнейшему распаду до конечных продуктов.
Схема обмена фенилаланина и тирозина
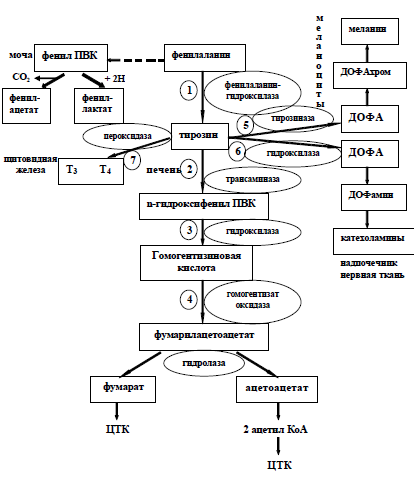
Схема обмена фенилаланина и тирозина
|
2. Нарушения обмена фенилаланина и тирозина |
| ||||||||||
|
Возможные нарушения обмена показаны на схеме обмена фени- | |||||||||||
|
лаланина и тирозина цифрами 1-7. |
|
|
|
|
|
|
| ||||
|
1. При отсутствии или недостаткефенилаланингидроксилазы | |||||||||||
|
(коферментом |
фенилаланингидроксилазы |
служит |
тетрагидробиопте | ||||||||
рин – ТГБП) развивается фенилкетонурия или фенилпировиноградная олигофрения. Это наследственное заболевание развивается с частотой 1 случай на 10 тыс. новорожденных (в Беларуси – 1:6 тыс. новорож-денных). Фенилаланин не может превратиться в тирозин и накаплива-ется в крови. Основная масса фенилаланина трансаминируется с обра-зованием фенилпирувата, который выделяется с мочой– фенилкето-нурия. С мочой также выводятся фениллактат,фенилацетат, фенил-ацетилглутамин, которые придают моче “запах мышей”.
В результате нарушения превращения фенилаланина в тирозин образуется дефицит тирозина (он становится незаменимой аминокис-лотой), дефицит катехоламинов, гормонов щитовидной железы, мела-нина. 80% больных – блондины со светлой кожей и голубыми глазами. Тяжелые проявления фенилкетонурии связаны с токсическим действи-ем на клетки мозга высоких концентраций фенилаланина,фенилпиру-вата и фениллактата. Большие концентрации фенилаланина ограничи-вают транспорт тирозина и триптофана через гематоэнцефалический барьер и тормозят синтез нейромедиаторов. Чувствительность нервной системы к токсическому влиянию продуктов обмена фенилаланина,к дефициту гормонов и медиаторов наиболее высока в раннем возрасте в период созревания мозга, поэтому развивается фенилпировиноград-ная олигофрения, отставание в психическом и физическом развитии.
Известны 2 формы фенилкетонурии:
Классическая – наследственное заболевание, связанное с мута-циями в гене фенилаланингидроксилазы. Наиболее тяжелые проявле-ния – нарушение умственного и физического развития,судорожный синдром.
Вариантная (коферментзависимая гиперфенилаланинемия) – следствие мутаций в генах, контролирующих метаболизм тетрагидро-биоптерина. Клинические проявления близки, но не во всем совпада-ют с классической формой. Тетрагидробиоптерин необходим для реакций гидроксилирова-ния не только фенилаланина, но и тирозина и триптофана, поэтому при недостатке этого кофермента нарушается метаболизм всех3 амино-кислот, в том числе синтез нейромедиаторов — катехоламинов и серо-тонина. Заболевание характеризуется тяжелыми неврологическими на-рушениями и ранней смертью («злокачественная» фенилкетонурия).
Ранняя диагностика фенилкетонурии осуществляется по про-грамме массового скрининга, основанной на применении теста Гатри. Это микробиологический тест, при котором диск фильтровальной бу-маги, содержащий кровь из пятки новорожденного, помещают на чаш-ку с посеянными микроорганизмамиBacillus Subtilis, нуждающимися для своего роста в фенилаланине, источником которого является обра-зец крови. Рост микроорганизмов определяется как положительный тест, указывающий на необходимость определения концентрации фе-нилаланина в крови. Однако более надежным для диагностики фенил-кетонурии является определение количества фенилаланина в крови, взятой из прокола кожи на пятке между6-10 днями жизни (накануне выписки из роддома). Концентрация фенилаланина в норме– 15мг/л, при заболевании увеличивается до 600 мг/л.
Рекомендуемая некоторыми авторами для диагностики фенилке-тонурии проба Фелинга (мочу новорожденных исследуют, добавляя в нее FeCl3, который в присутствии фенилпирувата дает оливково-зеленое окрашивание) дает положительный результат примерно через 6 недель после рождения, когда уже могут развиться необратимые из-менения головного мозга.
У недоношенных детей может быть повышенное содержание фе-нилаланина без фенилкетонурии вследствие замедленного созревания ферментных систем. Диф. диагноз – определение концентрации в кро-ви тирозина – при фенилкетонурии она не повышена, а повышена при других состояниях.
Лечение фенилкетонурии состоит в специальной диете, при ко-торой белок замещается смесью аминокислот с низким содержанием фенилаланина и достаточным содержанием тирозина. Ранее полагали, что пищевой контроль (безфенилаланиновая диета) необходим только в течение первых 10 лет жизни, однако современные данные говорят о необходимости пожизненного придерживания этой диеты.
В некоторых случаях даже раннее начало лечения не может пре-дотвратить умственные нарушения, но в этих случаях тяжесть их су-щественно меньшая, чем в отсутствие лечения.
Пациенты с фенилкетонурией должны воздерживаться от употребления каких-либо продуктов, содержащих аспартам – очень распространенный искусственный заменитель сахара.Эта пищевая добавка расщепляется в ЖКТ до фенилаланина. Особенно важным яв-ляется то, что все пищевые продукты, в том числе сладкие напитки, должны иметь указание на этикетке о содержании этого искусст-венного сахара.
2. Тирозинтрансаминаза печени – при ее недостаточности раз-вивается тирозинемия (синдром Рихнера-Ханхерта). В крови и моче повышается содержание тирозина, клинически отмечается поражение глаз, кожи, умеренная умственная отсталость.
Для нормальной активности п-оксифенилПВКгидроксилазы
необходим витамин С, больные цингой экскретируют с мочой непол-ностью окисленные продукты метаболизма тирозина. В ходе этой ре-акции происходит сочетанная миграция боковой цепи,декарбоксили-рование и гидроксилирование кольца.При недостаточности этого фермента в крови повышается содержание тирозина и фенилаланина, в моче – n-гидроксифенилпирувата. При лечении назначают бедную белком диету. Клинически обнаруживаются хронические дегенератив-ные изменения в печени и почках.
Гомогентизатоксидаза – это диоксигеназа, расщепляющая
ароматическое кольцо гомогентизата молекулой О. При недостаточности этого фермента в печени и крови накапливается гомогентизино-вая кислота. Она в больших количествах выводится с мочой. При стоя-нии мочи на воздухе гомогентизиновая кислота чернеет вследствие об-разования из нее темного пигмента – алкаптона, заболевание – алкап-тонурия. Никаких других проявлений заболевания в раннем возрасте не наблюдается. Постепенное накопление гомогентизиновой кислоты в тканях, особенно в хрящах и ее окисление приводит с возрастом к об-щей пигментации или охронозу (охра – коричневая) – темный кончик носа, ушей. У пожилых людей это явление сопровождается характер-ными поражениями крупных сосудов и позвоночника, артритом.
Частота заболевания – 2-5 случаев на 1 млн. новорожденных. 5. Медьсодержащая тирозиназа в клетках базального слоя эпидермиса, волосяных луковицах, в субстанции “нигра” мозга, в моз-говом веществе надпочечников, в глазу производит окисление тирози-на в ДОФА, который далее в меланобластах неферментативно образует пигмент меланин.
Потемнение кожи человека вызывается ультрафиолетовым облу-чением тирозина, приводящим к образованию ДОФА. Врожденное отсутствие тирозиназы приводит кальбинизму, который в зависимо-сти от степени нарушения проявляется или уменьшением пигментации или ее полным отсутствием. Клинически: нарушение остроты зрения, психопатия, фотобоязнь. Частота заболевания 1:20000.
6. В клетках надпочечников, нейронах окисление тирозина через стадию ДОФА приводит к образованиюдофамина, норадреналина,адреналина.Нарушение синтеза катехоламинов может вызывать различные нервно-психические заболевания, причём патологические отклонения наблюдаются как при снижении, так и при увеличении их количества.Болезнь Паркинсона
Заболевание развивается при недостаточности дофамина в чёр-ной субстанции мозга. Это одно из самых распространённых невроло-гических заболеваний (частота 1:200 среди людей старше 60 лет). При этой патологии снижена активность тирозингидроксилазы,ДОФА-декарбоксилазы. Заболевание сопровождается тремя основными сим-птомами: акинезия (скованность движений), ригидность (напряжение мышц), тремор (непроизвольное дрожание). Дофамин не проникает че-рез гематоэнцефалический барьер и как лекарственный препарат не используется. Для лечения паркинсонизма предлагаются следующие принципы:
Заместительная терапия препаратами-предшественниками дофамина (производными ДОФА) — леводопа, мадопар, наком и др. Подавление инактивации дофамина ингибиторами МАО (депренил, ниаламид, пиразидол и др.). Депрессивные состояния часто связаны со снижением в нерв-ных клетках содержания дофамина и норадреналина. Гиперсекреция дофамина в височной доле мозга наблюдается при шизофрении. 7. В щитовидной железе из тирозина образуются гормоны 3Ти Т4. Тиреоидные гормоны синтезируются из тирозина в ходе ряда реак-ций, инициируемых тиреоидпероксидазой. При этом процессе неор-ганический йод окисляется в присутствии перекиси водорода,и затем йодид замещает атом водорода в тирозине.Дефицит тиреоидной пе-роксидазы – одна из причин зоба и гипотиреоидизм. 8. ПИЩЕВЫЕ ЖИРЫ: ПЕРЕВАРИВАНИЕ, ВСАСЫВАНИЕ ПРОДУКТОВ РАСЩЕПЛЕ-НИЯ. РОЛЬ ЖЕЛЧНЫХ КИСЛОТ. ТРАНСПОРТ ЛИПИДОВ В ОРГАНИЗМЕ.
Жиры – это гидрофобные продукты, ферменты их переваривающие – гидрофильны. Поэтому для увеличения поверхности соприкосновения с гидрофильными ферментами жиры должны эмульгироваться (разбиться на капли). Эмульгирование жиров может происходить толь-ко в кишечнике, поэтому основное место переваривания жиров – тонкий кишечник (duodenum).
Факторы эмульгирования: 1) перистальтика кишечника, 2) пробулькивание через содержимое кишечника пузырьков СО, образующегося при нейтролизации кислого содержимого желудка бикарбонатами поджелудочного сока, 3) действие поверхностно-активных веществ– желчных кислот, пептонов (продукты переваривания белков) и др.
Основную массу жира пищи составляют триацилглицерины. В их переваривании участвует поджелудочная липаза. Она попадает в кишечник в неактивном виде и активируется колипазой и желчными кислотами, оптимум рН ее действия – слабощелочная среда. Продукты переваривания триацилглицеринов: глицерин, жирные кислоты, диацилглицерины, моноацилглицерины.
У новорожденных в желудке слабокислая среда,они питаются эмульгированным жиром молока, поэтому у грудных детей возможно переваривание ТГ в желудке под действием желудочной липазы.
Гидролиз фосфолипидов происходит в кишечнике под действием фосфолипаз 4-х типов:
|
|
|
|
|
|
|
|
A1 |
O |
|
|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
CH2 |
|
|
|
|
O |
|
|
|
C |
|
|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
R |
|
|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
A2 |
1 |
|
|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
O |
|
|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
O |
|
|
|
|
C |
|
|
|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
CH |
|
|
|
|
|
|
|
|
|
R |
|
|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
|
С |
|
|
|
|
|
|
|
2 |
Д |
|
|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
OH |
|
CH N+(CH ) |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
O |
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
CH2 |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
P |
|
|
O |
|
CH |
|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2 |
3 3 |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
O
Фосфолипаза А1 – отщепляет жирную кислоту в 1-ом положении
фосфолипаза А2 – ненасыщенную жирную кислоту во2-ом, при этом образуется лизофосфолипиды, которые при попадании в кровь могут вызывать гемолиз эритроцитов.
Фосфолипаза С отщепляет фосфорилированный спирт
фосфолипаза Д - свободный спирт.
Итак, конечные продукты переваривания фосфолипидов: глицерин, жирные кислоты, Н3РО4 и азотистые спирты: холин, этаноламин, серин, инозит.
Поступающие с пищей эфиры холестерина расщепляются панкреатической холестеролэстеразой на холестерин и жирную кислоту.
В результате переваривания жиров в кишечнике образуется гидрофобные продукты переваривания(холестерин, жирные кислоты с числом С атомов более12, ди- и моноацилглицерины и гидрофильные (глицерин, короткие жирные кислоты, Н3РО4, холин, серин, этаноламин и др.). Всасывание гидрофильных продуктов переваривания в стенку слизистой кишечника происходит самостоятельно, а гидрофобные продукты всасываются в составе мицелл. Большую роль в образовании мицелл играют желчные кислоты. Мицелла – это сферический комплекс, в центре которого находятся транспортируемые гидрофобные продукты переваривания, окруженные желчными кислотами. Придя в слизистую кишечника, мицелла разгружается. Желчные кислоты могут попасть дальше в воротную вену ® в печень ® желчь ® кишечник. Это рециркуляция желчных кислот. Однако основная масса желчных кислот удаляется с содержимым кишечника.
Роль желчных кислот в переваривании жиров:
1) эмульгируют жиры, 2) активируют липазу, 3) создают оптимум рН для действия липазы 4) участвуют во всасывании гидрофобных про-дуктов переваривания, образуя мицеллы.
Различают экзогенный, т.е. транспорт пищевых липидов и эндогенный транспорт – т.е. липидов, синтезированных в организме.
Экзогенный транспорт.
Ресинтезированные в энтероцитах триацилглицерины вместе с фосфолипидами, холестерином и белками включаются в хиломикроны. Хиломикроны содержат апопротеин В48 и апоА. Хиломикроны из энте-роцитов попадают в грудной лимфатический проток и далее– в кровь, здесь они встречаются с частицами ЛПВП, содержащими апоЕ и апоС. ХМ отдают апоА частицам ЛПВП, а взамен приобретают апоЕ и апоС. Этот обмен очень важен, т.к. апоСII служит активатором фермента ли-попротеинлипазы – ЛПЛ. Этот фермент синтезируется и секретируется клетками жировой и мышечной ткани, клетками молочных желез. Сек-ретируемый фермент прикрепляется к плазматической мембране эндо-телиальных клеток капилляров тех тканей,где он синтезировался. АпоСII, находящейся на поверхности ХМ, активирует ЛПЛ, она гидро-лизует триацилглицерины в составе ХМ до глицерина и жирных кислот.
Освободившиеся жирные кислоты либо поступают в клетки жи-ровой и мышечной ткани, либо соединяются с альбуминами плазмы и транспортируются в общий кровоток. В результате действия ЛПЛ ХМ резко уменьшаются в размерах и называются уже ремнантами (ремнант-остаток). Ремнанты ХМ рецепторным путем захватываются печенью и с ними в печень попадают в основном холестерин и небольшое количест-во триацилглицеринов. Клетки печени включают поступивший холес рин, а также вновь синтезированный, триацилглицерины, фосфолипиды и белки в состав ЛПОНП.
Эндогенный транспорт.
Основными белками ЛПОНП являются апоВ и апоС, а липидами – триацилглицерины. Кроме того, в печени синтезируется ещё один класс липопротеинов-ЛПВП,у них основной белок – апоА, много фосфолипидов и свободного холестерина, а ядро пустое – так называемые насцентные ЛПВП. Они играют большую роль в обратном транспорте холестерина из клеток периферических тканей в печень. Т.к. ЛПОНП содержат апоСII, происходит активация ЛПЛ, которая гидролизует триацилглицерины ЛПОНП и превращает ЛПОНП в липопротеины промежуточной плотности ЛППП. ЛППП под действием фермента, синтезируемого в печени и секретируемого в кровь,– печеночной триацилглицеринлипазы, превращаются в ЛПНП. Основным липидом в ЛПНП становится холестерин, который в составе ЛПНП переносится к клеткам всех тканей. ЛПНП образуются непосредственно в сосудистом русле и участвуют в прямом транспорте холестерина.
9. ВАЖНЕЙШИЕ ЛИПИДЫ ТКАНЕЙ ЧЕЛОВЕКА. ФУНКЦИИ ЛИПИДОВ. ДЕПОНИРО-ВАНИЕ И МОБИЛИЗАЦИЯ ЖИРОВ В ЖИРОВОЙ ТКАНИ. РЕГУЛЯЦИЯ. ТРАНСПОРТ И ИСПОЛЬЗОВАНИЕ ЖИРНЫХ КИСЛОТ. ХИМИЗМ И ЭНЕРГЕТИКА ОКИСЛЕНИЯ ГЛИЦЕРИНА. Общая характеристика липидов.
Липиды – это обширная группа соединений, различающихся по химической структуре и выполняемым функциям. Признаки липидов: 1) нерастворимость в воде, 2) растворимость в эфире, хлороформе, бензоле, т.е. в полярных органических растворителях3) содержание в своем составе жирных кислот.
По своему строению липиды, в большинстве случаев, это сложные эфиры высших жирных кислот с глицерином или некоторыми другими спиртами, также в состав могут входить фосфорная кислота, азотистые основания, углеводы.
Функции липидов
1. Липиды в виде комплекса с белками являются структурными элементами клеточных мембран. Они определяют текучесть мембраны и транспорт веществ в клетку.
2. Липиды служат энергетическим материалом для организма. При окислении 1 г. жира выделяется 39 кДж энергии, что в 2 раза больше, чем при окислении 1 г углевода.
3.В виде жировой прокладки предохраняют тело и органы животных от механического повреждения (жировая капсула у почек, сальник защищает органы брюшной полости).
4.Липиды сохраняют тепло в организме, обладая хорошими термоизоляционными свойствами (морские животные, пловцы-моржи).
5.Эйкозаноиды обладают регуляторной активностью.
6.Гликолипиды являются важными компонентами нервной ткани, оказывая существенное влияние на функционирование нервной системы.
7.Некоторые липиды являются предшественниками многих биологически активных веществ. Например, из фосфолипидов образуются простагландины, тромбоксан, простациклин, диацилглицерол, инози-толтрифосфат и т.д. Холестерин служит предшественником желчных кислот, стероидных гормонов надпочечников, семенников, яичников и плаценты, из него образуется витамин Д.
8. Эстетическая роль - жировая прокладка смягчает контуры скелета, образуя «округлости», которые создают привычный образ тела человека.
С нарушениями обмена липидов связаны такие грозные заболевания как атеросклероз, ожирение, желчно-каменная болезнь и другие.
Жиры – это гидрофобные продукты, ферменты их переваривающие – гидрофильны. Поэтому для увеличения поверхности соприкосновения с гидрофильными ферментами жиры должны эмульгироваться (разбиться на капли). Эмульгирование жиров может происходить только в кишечнике, поэтому основное место переваривания жиров – тонкий кишечник.
Основную массу жира пищи составляют триацилглицерины. В их переваривании участвует поджелудочная липаза. Она попадает в кишечник в неактивном виде и активируется колипазой и желчными кислотами, оптимум рН ее действия – слабощелочная среда. Продукты переваривания триацилглицеринов: глицерин, жирные кислоты, диацилглицерины, моноацилглицерины.
Синтез жиров в жировой ткани
В жировой ткани для синтеза жиров используются в основном жирные кислоты, освободившиеся при гидролизе жиров ХМ и ЛПОНП. Жирные кислоты поступают в адипоциты, превращаются в производные КоА и взаимодействуют с глицерол-3-фосфатом, образуя сначала лизофосфатидную кислоту, а затем фосфатидную. Фосфатидная кислота после дефосфорилирования превращается в диацилглицерол, который ацилируется с образованием триацилглицерола.
Кроме жирных кислот, поступающих в адипоциты из крови, в этих клетках идёт и синтез жирных кислот из продуктов распада глюкозы. В адипоцитах для обеспечения реакций синтеза жира распад глюкозы идёт по двум путям: гликолиз, обеспечивающий образование глицерол-3-фосфата и ацетил-КоА, и пентозофосфатный путь, окислительные реакции которого обеспечивают образование NADPH, служащего донором водорода в реакциях синтеза жирных кислот.
Молекулы жиров в адипоцитах объединяются в крупные жировые капли, не содержащие воды, и поэтому являются наиболее компактной формой хранения топливных молекул.
Синтез ТАГ в печени. Образование ЛПОНП в печени и транспорт жиров в другие ткани
Печень - основной орган, где идёт синтез жирных кислот из продуктов гликолиза. В гладком ЭР гепатоцитов жирные кислоты активируются и сразу же используются для синтеза жиров, взаимодействуя с глицерол-3-фосфатом. Как и в жировой ткани, синтез жиров идёт через образование фосфатидной кислоты. Синтезированные в печени жиры упаковываются в ЛПОНП и се-ретируются в кровь.
ЛПОНП из печени секретируются в кровь, где на них, как и на ХМ, действует ЛП-липаза. Жирные кислоты поступают в ткани, в частности в адипоциты, и используются для синтеза жиров. В процессе удаления жиров из ЛПОНП под действием ЛП-липазы ЛПОНП сначала превращаются в ЛГШП, а затем в ЛПНП. В ЛПНП основными липидными компонентами служат холестерол и его эфиры, поэтому ЛПНП являются липопротеинами, доставляющими холестерол в периферические ткани. Глицерол, освободившийся из липопротеинов, кровью транспортируется в печень, где опять может использоваться для синтеза жиров.
Различают экзогенный, т.е. транспорт пищевых липидов и эндогенный транспорт – т.е. транспорт липидов, синтезированных в организме. э
Экзогенный транспорт
Ресинтезированные в энтероцитах триацилглицерины вместе с фосфолипидами, холестерином и белками включаются в хиломикроны. Хиломикроны содержат апопротеин В48 и апоА. Хиломикроны из энтероцитов попадают в грудной лимфатический проток и далее– в кровь, здесь они встречаются с частицами ЛПВП, содержащими апоЕ и апоС. ХМ отдают апоА частицам ЛПВП, а взамен приобретают апоЕ и апоС. Этот обмен очень важен, т.к. апоСII служит активатором фермента ли-попротеинлипазы – ЛПЛ. Этот фермент синтезируется и секретируется клетками жировой и мышечной ткани, клетками молочных желез. Сек-ретируемый фермент прикрепляется к плазматической мембране эндо-телиальных клеток капилляров тех тканей,где он синтезировался. АпоСII, находящейся на поверхности ХМ, активирует ЛПЛ, она гидро-лизует триацилглицерины в составе ХМ до глицерина и жирных кислот.
Освободившиеся жирные кислоты либо поступают в клетки жировой и мышечной ткани, либо соединяются с альбуминами плазмы и транспортируются в общий кровоток. В результате действия ЛПЛ ХМ резко уменьшаются в размерах и называются уже ремнантами (ремнант-остаток). Ремнанты ХМ рецепторным путем захватываются печенью и с ними в печень попадают в основном холестерин и небольшое количест-во триацилглицеринов. Клетки печени включают поступивший холестерин, а также вновь синтезированный, триацилглицерины, фосфолипиды и белки в состав ЛПОНП.
Эндогенный транспорт. Основными белками ЛПОНП являются апоВ и апоС, а липидами – триацилглицерины. Кроме того, в печени синтезируется ещё один класс липопротеинов-ЛПВП,у них основной белок – апоА, много фосфолипидов и свободного холестерина, а ядро пустое – так называемые насцентные ЛПВП. Они играют большую роль в обратном транспорте холестерина из клеток периферических тканей в печень. Т.к. ЛПОНП содержат апоСII, происходит активация ЛПЛ, ко-торая гидролизует триацилглицерины ЛПОНП и превращает ЛПОНП в липопротеины промежуточной плотности ЛППП. ЛППП под действием фермента, синтезируемого в печени и секретируемого в кровь,– пече-ночной триацилглицеринлипазы, превращаются в ЛПНП. Основным липидом в ЛПНП становится холестерин, который в составе ЛПНП пе-реносится к клеткам всех тканей. ЛПНП образуются непосредственно в сосудистом русле и участвуют в прямом транспорте холестерина.
Доказано, что синтез ЛПЛ происходит под влиянием инсулина. При сахарном диабете, когда отмечается дефицит инсулина,уровень ЛПЛ снижается. В результате в крови накапливается большое количест-во липопротеинов, богатых триацилглицеринами (IV тип ГЛП).