

5.4. Биосинтез пиримидиновых нуклеотидов.
5.4.1. Структура пиримидинового ядра проще, чем структура пуринового, и путь биосинтеза его короче.
Главное отличие от биосинтеза пуринов заключается в том, что сборка пиримидинового ядра не требует участия фосфорибозилпирофосфата (ФРПФ). Пиримидиновая структура сначала образуется, а потом взаимодействует с ФРПФ с образованием нуклеотидов.
Предшественниками атомов углерода и азота пиримидинового кольца являются СО2 и аминокислоты глутамин и аспартат (рисунок 5.6).
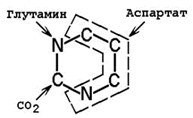
Рисунок 5.6. Происхождение атомов пиримидинового ядра.
5.4.2. При взаимодействии СО2 и глутамина образуется карбамоилфосфат. Реакция протекает в цитозоле с затратой АТФ.
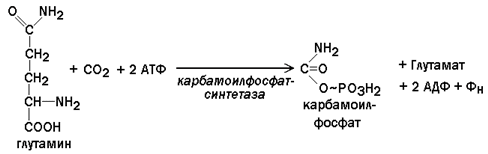
Следует напомнить, что карбамоилфосфат также образуется в процессе синтеза мочевины, но эта реакция происходит только в митохондриях печени и катализируется другим ферментом.
Карбамоилфосфатсинтетаза, участвующая в биосинтезе пиримидинов, - аллостерический фермент, он ингибируется УТФ, пуриновыми нуклеотидами, активируется ФРПФ.
5.4.3. Следующая реакция биосинтеза пиримидинов – образование карбамоиласпартата. Реакцию катализирует аспартат-транскарбамоилаза (АТК-аза).

АТК-аза – аллостерический фермент, его ингибитором является цитидинтрифосфат (ЦТФ) – конечный продукт биосинтеза пиримидиновых нуклеотидов.
Дальнейший путь образования пиримидиновых нуклеотидов представлен на рисунке 5.7.

Рисунок 5.7. Схема синтеза пиримидиновых нуклеотидов.
5.5. Особенности биосинтеза дезоксирибонуклеотидов
5.5.1. Дезоксирибонуклеотиды входят в состав ДНК. Содержание дезоксирибонуклеотидов в клетке обычно низкое и повышается только перед репликацией ДНК.
Дезоксирибонуклеотиды образуются в результате восстановления рибонуклеотидов. Эта реакция катализируется рибонуклеотидредуктазой (рисунок 5.8).
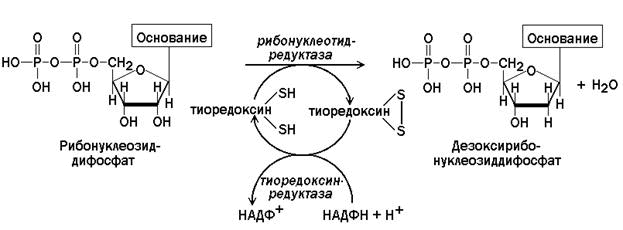
Рисунок 5.8. Схема образования дезоксирибонуклеотидов.
В процессе восстановления рибонуклеозиддифосфатов окисляется донор электронов – тиоредоксин. Окисленный тиоредоксин восстанавливается тиоредоксинредуктазой за счёт восстановленного НАДФ.
5.5.2. Тимидинмонофосфат образуется из дезоксиуридинмонофосфата при участии фермента тимидилатсинтазы. Источником метильной группы в реакции служит 5,10-метилен-тетрагидрофолиевая кислота (5,10-метилен-ТГФК), которая окисляется до дигидрофолиевой. Тетрагидрофолиевая кислота регенерируется дигидрофолатредуктазой в реакции, которая требует участия НАДФН. Образование 5,10-метилен-ТГФК происходит в реакции ТГФК с серином или глицином (см. тему 3).
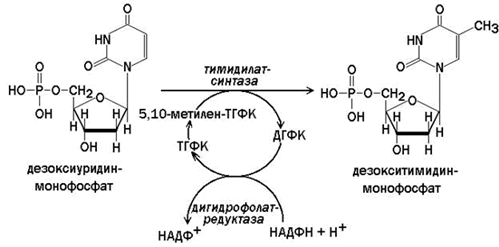
Рисунок 5.9. Схема образования тимидилового нуклеотида.
5.6. Нарушения обмена нуклеотидов.
5.6.1. При нарушениях пуринового обмена часто наблюдается гиперурикемия – повышение содержания мочевой кислоты в крови. Гиперурикемия может быть первичной или вторичной.
Первичная гиперурикемия является ведущим симптомом подагры – полиэтиологического заболевания, как правило, наследственной природы. Гиперурикемия при подагре обусловлена главным образом, избыточным образованием образованием мочевой кислоты, а также снижением её экскреции с мочой. Значительная и длительная гиперурикемия сопровождается отложением солей мочевой кислоты в хрящевой ткани, сухожилиях и слизистых сумках суставов. Накопление кристаллов уратов в тканях может вызывать резкую воспалительную реакцию (подагрический артрит), что приводит впоследствии к деформации сустава. Избыток мочевой кислоты способствует также образованию уратных камней в нижних отделах мочевыводящих путей.
Повышение уровня мочевой кислоты в крови отмечается также при наследственных дефектах некоторых ферментов:
Синдром Леша-Нихана (полное отсутствие ГГФРТ) наследуется как сцепленный с Х-хромосомой рецессивный признак. Болезнь характеризуется параличом, сопровождающимся судорогами, стремлением к членовредительству и тяжёлой гиперурикемией. Вследствие ферментативного дефекта нарушается переход гуанина и гипоксантина в ГМФ и ИМФ соответственно и указанные пуриновые основания превращаются в мочевую кислоту. Кроме того, повышенная концентрация ФРПФ способствует усилению синтеза пуринов de novo. Биохимическая основа неврологических отклонений при синдроме Леша-Нихана неизвестна.
Гликогеноз I типа или болезнь Гирке (дефицит глюкозо-6-фосфатазы) сопровождается повышением активности пентозофосфатного пути и приводит к повышению внутриклеточного уровня рибозо-5-фосфата, из которого синтезируется ФРПФ. Повышенный уровень ФРПФ приводит к увеличению синтеза пуринов de novo. Для данного заболевания характерен также лактатный ацидоз, приводящий к повышению порога секреции уратов почками; это способствует накоплению уратов в организме.
Вторичная гиперурикемия сопутствует заболеваниям, сопровождающимся усиленным распадом клеток (лейкозы, серповидно-клеточная анемия, сахарный диабет, псориаз).
5.6.2. Реже встречается гипоурикемия – снижение содержания мочевой кислоты в крови. Она может быть связана с понижением реабсорбции уратов из клубочкового фильтрата в почках. В этом случае наблюдается увеличение экскреции мочевой кислоты с мочой.
Гипоурикемия развивается и при недостаточности ксантиноксидазы, возникающей при генетическом дефекте фермента или при тяжёлом поражении печени. Это состояние сопровождается повышенной экскрецией гипоксантина и ксантина (ксантинурией), а также образованием в почках ксантиновых камней.
5.6.3. Описаны два иммунодефицитных заболевания, связанные с недостаточностью ферментов метаболизма пуринов. Недостаточность аденозиндезаминазы сопровождается снижением количества и нарушением функции как тимусных лимфоцитов (Т-клеток), так и лимфоцитов костного мозга (В-клеток). При недостаточности пуриннуклеозид-фосфорилазы функции В-клеток остаются нормальными, но значительно нарушаются функции Т-клеток. Метаболические нарушения при данных заболеваниях связаны с накоплением дезоксирибонуклеозидтрифосфатов (дГТФ и дАТФ), которые аллостерически ингибируют рибонуклеотидредуктазу. Это, в свою очередь, приводит к снижению содержания в Т-лимфоцитах предшественников синтеза ДНК, главным образом дЦТФ. Таким образом, Т-клетки размножаться не могут.