

- •Overview
- •Preface
- •Translator’s Note
- •Contents
- •1. Fundamentals
- •Microscopic Anatomy of the Nervous System
- •Elements of Neurophysiology
- •Elements of Neurogenetics
- •General Genetics
- •Neurogenetics
- •Genetic Counseling
- •2. The Clinical Interview in Neurology
- •General Principles of History Taking
- •Special Aspects of History Taking
- •3. The Neurological Examination
- •Basic Principles of the Neurological Examination
- •Stance and Gait
- •Examination of the Head and Cranial Nerves
- •Head and Cervical Spine
- •Cranial Nerves
- •Examination of the Upper Limbs
- •Motor Function and Coordination
- •Muscle Tone and Strength
- •Reflexes
- •Sensation
- •Examination of the Trunk
- •Examination of the Lower Limbs
- •Coordination and Strength
- •Reflexes
- •Sensation
- •Examination of the Autonomic Nervous System
- •Neurologically Relevant Aspects of the General Physical Examination
- •Neuropsychological and Psychiatric Examination
- •Psychopathological Findings
- •Neuropsychological Examination
- •Special Considerations in the Neurological Examination of Infants and Young Children
- •Reflexes
- •4. Ancillary Tests in Neurology
- •Fundamentals
- •Imaging Studies
- •Conventional Skeletal Radiographs
- •Computed Tomography (CT)
- •Magnetic Resonance Imaging (MRI)
- •Angiography with Radiological Contrast Media
- •Myelography and Radiculography
- •Electrophysiological Studies
- •Fundamentals
- •Electroencephalography (EEG)
- •Evoked potentials
- •Electromyography
- •Electroneurography
- •Other Electrophysiological Studies
- •Ultrasonography
- •Other Ancillary Studies
- •Cerebrospinal Fluid Studies
- •Tissue Biopsies
- •Perimetry
- •5. Topical Diagnosis and Differential Diagnosis of Neurological Syndromes
- •Fundamentals
- •Muscle Weakness and Other Motor Disturbances
- •Sensory Disturbances
- •Anatomical Substrate of Sensation
- •Disturbances of Consciousness
- •Dysfunction of Specific Areas of the Brain
- •Thalamic Syndromes
- •Brainstem Syndromes
- •Cerebellar Syndromes
- •6. Diseases of the Brain and Meninges
- •Congenital and Perinatally Acquired Diseases of the Brain
- •Fundamentals
- •Special Clinical Forms
- •Traumatic Brain injury
- •Fundamentals
- •Traumatic Hematomas
- •Complications of Traumatic Brain Injury
- •Intracranial Pressure and Brain Tumors
- •Intracranial Pressure
- •Brain Tumors
- •Cerebral Ischemia
- •Nontraumatic Intracranial Hemorrhage
- •Infectious Diseases of the Brain and Meninges
- •Infections Mainly Involving the Meninges
- •Infections Mainly Involving the Brain
- •Intracranial Abscesses
- •Congenital Metabolic Disorders
- •Acquired Metabolic Disorders
- •Diseases of the Basal Ganglia
- •Fundamentals
- •Diseases Causing Hyperkinesia
- •Other Types of Involuntary Movement
- •Cerebellar Diseases
- •Dementing Diseases
- •The Dementia Syndrome
- •Vascular Dementia
- •7. Diseases of the Spinal Cord
- •Anatomical Fundamentals
- •The Main Spinal Cord Syndromes and Their Anatomical Localization
- •Spinal Cord Trauma
- •Spinal Cord Compression
- •Spinal Cord Tumors
- •Myelopathy Due to Cervical Spondylosis
- •Circulatory Disorders of the Spinal Cord
- •Blood Supply of the Spinal Cord
- •Arterial Hypoperfusion
- •Impaired Venous Drainage
- •Infectious and Inflammatory Diseases of the Spinal Cord
- •Syringomyelia and Syringobulbia
- •Diseases Mainly Affecting the Long Tracts of the Spinal Cord
- •Diseases of the Anterior Horns
- •8. Multiple Sclerosis and Other Myelinopathies
- •Fundamentals
- •Myelin
- •Multiple Sclerosis
- •Other Demyelinating Diseases of Unknown Pathogenesis
- •9. Epilepsy and Its Differential Diagnosis
- •Types of Epilepsy
- •Classification of the Epilepsies
- •Generalized Seizures
- •Partial (Focal) Seizures
- •Status Epilepticus
- •Episodic Neurological Disturbances of Nonepileptic Origin
- •Episodic Disturbances with Transient Loss of Consciousness and Falling
- •Episodic Loss of Consciousness without Falling
- •Episodic Movement Disorders without Loss of Consciousness
- •10. Polyradiculopathy and Polyneuropathy
- •Fundamentals
- •Polyradiculitis
- •Cranial Polyradiculitis
- •Polyradiculitis of the Cauda Equina
- •Polyneuropathy
- •Fundamentals
- •11. Diseases of the Cranial Nerves
- •Fundamentals
- •Disturbances of Smell (Olfactory Nerve)
- •Neurological Disturbances of Vision (Optic Nerve)
- •Visual Field Defects
- •Impairment of Visual Acuity
- •Pathological Findings of the Optic Disc
- •Disturbances of Ocular and Pupillary Motility
- •Fundamentals of Eye Movements
- •Oculomotor Disturbances
- •Supranuclear Oculomotor Disturbances
- •Lesions of the Nerves to the Eye Muscles and Their Brainstem Nuclei
- •Ptosis
- •Pupillary Disturbances
- •Lesions of the Trigeminal Nerve
- •Lesions of the Facial Nerve
- •Disturbances of Hearing and Balance; Vertigo
- •Neurological Disturbances of Hearing
- •Disequilibrium and Vertigo
- •The Lower Cranial Nerves
- •Accessory Nerve Palsy
- •Hypoglossal Nerve Palsy
- •Multiple Cranial Nerve Deficits
- •12. Diseases of the Spinal Nerve Roots and Peripheral Nerves
- •Fundamentals
- •Spinal Radicular Syndromes
- •Peripheral Nerve Lesions
- •Fundamentals
- •Diseases of the Brachial Plexus
- •Diseases of the Nerves of the Trunk
- •13. Painful Syndromes
- •Fundamentals
- •Painful Syndromes of the Head And Neck
- •IHS Classification of Headache
- •Approach to the Patient with Headache
- •Migraine
- •Cluster Headache
- •Tension-type Headache
- •Rare Varieties of Primary headache
- •Symptomatic Headache
- •Painful Syndromes of the Face
- •Dangerous Types of Headache
- •“Genuine” Neuralgias in the Face
- •Painful Shoulder−Arm Syndromes (SAS)
- •Neurogenic Arm Pain
- •Vasogenic Arm Pain
- •“Arm Pain of Overuse”
- •Other Types of Arm Pain
- •Pain in the Trunk and Back
- •Thoracic and Abdominal Wall Pain
- •Back Pain
- •Groin Pain
- •Leg Pain
- •Pseudoradicular Pain
- •14. Diseases of Muscle (Myopathies)
- •Structure and Function of Muscle
- •General Symptomatology, Evaluation, and Classification of Muscle Diseases
- •Muscular Dystrophies
- •Autosomal Muscular Dystrophies
- •Myotonic Syndromes and Periodic Paralysis Syndromes
- •Rarer Types of Muscular Dystrophy
- •Diseases Mainly Causing Myotonia
- •Metabolic Myopathies
- •Acute Rhabdomyolysis
- •Mitochondrial Encephalomyopathies
- •Myositis
- •Other Diseases Affecting Muscle
- •Myopathies Due to Systemic Disease
- •Congenital Myopathies
- •Disturbances of Neuromuscular Transmission−Myasthenic Syndromes
- •15. Diseases of the Autonomic Nervous System
- •Anatomy
- •Normal and Pathological Function of the Autonomic Nervous System
- •Sweating
- •Bladder, Bowel, and Sexual Function
- •Generalized Autonomic Dysfunction
- •Index

154 7 Diseases of the Spinal Cord
few days). The intrinsic muscle reflexes are diminished because of posterior root involvement, pyramidal tract signs are common, and mental abnormalities may be seen, ranging to dementia. The diagnosis rests on the demonstration of vitamin B12 deficiency. Vitamin B12 must be given as rapidly as possible, preferably by the intramuscular route.
Long tracts of the spinal cord can also be involved in paraneoplastic syndromes, tabes dorsalis due to neurosyphilis (pp. 116 f.), adrenoleukodystrophy (p. 122), and a number of congenital metabolic diseases.
Diseases of the Anterior Horns
Isolated diseases of the anterior horn cells are mostly of genetic (spinal muscular atrophies) or infectious origin (acute anterior poliomyelitis, p. 150). Amyotrophic lateral sclerosis, in which there is simultaneous degeneration of the anterior horn cells and the corticospinal and/or corticobulbar tracts, is usually sporadic.
The typical clinical features of diseases involving chronic loss of anterior horn cells are summarized in Table 7.3. Some of these diseases are described in further detail in this section.
Spinal Muscular Atrophies
These diseases are due to a genetic defect on chromosome 5 that causes isolated degeneration of the second (lower) motor neurons, i. e., the motor neurons of the anterior horn cells and the cranial nerve nuclei. The result is the typical clinical syndrome of anterior horn degeneration described above (flaccid weakness, muscle atrophy, loss of reflexes, fasciculations). The main clinical types of spinal muscular atrophy are classified according to their age of onset and the pattern of motor deficits that they cause:
In the early infantile type (Werdnig−Hoffmann), neonates and infants suffer from rapidly progressive muscle weakness, beginning in the muscles of the pelvic girdle. The affected children can survive for no more than a few years.
Table 7.3 Diseases with chronic involvement of the anterior horn ganglion cells*
Disease |
Affected structures |
Symptoms and signs |
Remarks |
Etiology |
|
|
|
|
|
Infantile spinal muscular |
anterior horn ganglion |
muscle atrophy and |
affects infants and small |
autosomal recessive in- |
atrophy |
cells of the spinal cord |
weakness, hypotonia, |
children; rapidly fatal |
heritance (?); gene on |
(Werdnig−Hoffmann) |
(and bulbar motor neu- |
fasciculations of the |
|
chromosome 5 |
|
rons) |
tongue |
|
|
Pseudomyopathic spinal |
anterior horn ganglion |
muscle atrophy and fas- |
children and adoles- |
irregular dominance; |
muscular atrophy |
cells of the spinal cord |
ciculations, progressive |
cents, usually begins in |
gene on chromosome 5 |
(Kugelberg−Welander) |
|
gait disturbance, no |
the lower limbs, slowly |
|
|
|
bulbar involvement |
progressive |
|
Adult spinal muscular |
anterior horn ganglion |
muscle atrophy, weak- |
younger adults; begins |
usually isolated, of un- |
atrophy |
cells of the spinal cord |
ness, and fasciculations |
distally (hands) |
known etiology; occasio- |
(Aran−Duchenne) |
|
|
|
nally due to syphilis |
Proximal spinal muscular |
anterior horn ganglion |
muscle atrophy, weak- |
adults; slowly progres- |
unknown; occasionally |
atrophy of the shoulder |
cells of the spinal cord |
ness, and fasciculations |
sive |
due to syphilis |
girdle (Vulpian−Bernhardt) |
|
in the shoulder girdle |
|
|
|
|
region |
|
|
Amyotrophic lateral scle- |
anterior horn ganglion |
muscle atrophy and |
rosis (sometimes including |
cells of the spinal cord, |
weakness, fasciculations, |
true bulbar palsy) |
perhaps also motor cra- |
bulbar palsy with dys- |
|
nial nerve nuclei, pyrami- |
arthria and dysphagia, |
|
dal tracts, and cortico- |
spasticity, pyramidal |
|
bulbar tracts |
tract signs |
adults, rapidly progres- |
usually sporadic, rarely |
sive and lethal; juvenile |
familial |
(familial) cases are less |
|
common and have a re- |
|
latively benign course |
|
*A number of rarer neurological diseases affect the anterior horn ganglion cells as one component of a wider disease process; these include Creutzfeldt−Jakob disease, orthostatic hypotension, diabetic amyotrophy (?), metacarcinomatous myelopathy, organic mercury poisoning, and others.
Mumenthaler / Mattle, Fundamentals of Neurology © 2006 Thieme All rights reserved. Usage subject to terms and conditions of license.
Diseases of the Anterior Horns 155
Pseudomyopathic spinal muscular atrophy (Kugelberg−Welander) becomes symptomatic between the 2nd and 10th years of life. The pelvic girdle is most severely affected at first, as in the early infantile type, but the weakness and atrophy progress more slowly, and the overall prognosis is much more favorable. The first signs of disease are progressive quadriceps weakness, disappearance of the patellar tendon reflex, and, sometimes, pseudohypertrophy of the calves.
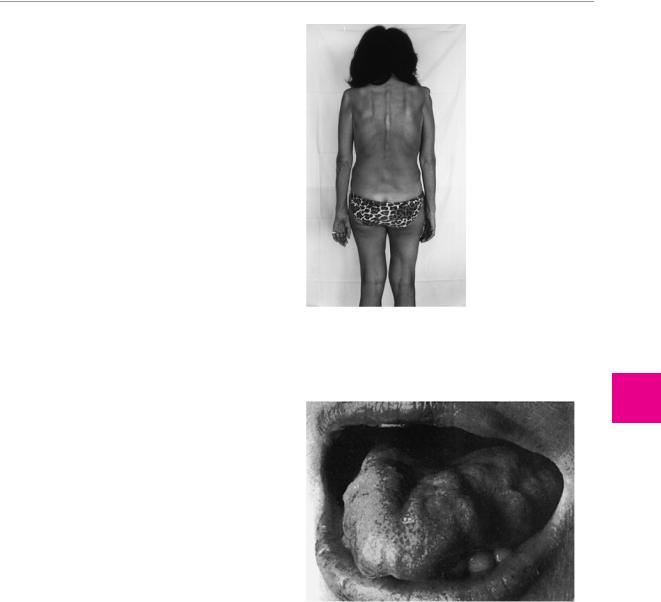
Types that become symptomatic from the third decade onward tend to be generalized, though the initial presentation tends to be either mainly distal (Aran− Duchenne) or mainly proximal (Vulpian−Bernhardt). The Aran−Duchenne type often presents with atrophy of the intrinsic muscles of the hand, the Vulpian− Bernhardt type with scapulohumeral atrophy. The latter is now considered a subtype of familial amyotrophic lateral sclerosis (see below); it affects not only the muscles of the limbs, but also those of the trunk and respiratory apparatus (Fig. 7.14).
Amyotrophic Lateral Sclerosis (ALS)
This disease, also known as motor neuron disease (MND), is characterized by combined degeneration of the first and second motor neurons. Its clinical features are thus a combination of flaccid paresis, muscular atrophy, and spasticity.
Epidemiology. Three-quarters of patients are men, most of them between the ages of 40 and 65. More than 95 % of cases are sporadic; the rare familial cases are thought to be due to a defect of the Cu/Zn superoxide dismutase gene.
Neuropathological hallmark of this disease is loss of anterior horn cells, combined with degeneration of the pyramidal and corticobulbar tracts and of the Betz pyramidal cells of the precentral gyri.
Characteristic clinical manifestations are:
weakness and atrophy of the muscle groups of the limbs and trunk (including the respiratory apparatus) and/or the bulbar muscles (tongue, throat), progressing slowly over months,
fasciculations,
exaggerated reflexes,
(in some patients) pyramidal tract signs,
intact sensation,
often muscle cramps and pain.
Course. At first, there is circumscribed, asymmetrical, predominantly distal muscle atrophy, which is usually most obvious in the intrinsic muscles of the hands. There may be accompanying pain or fasciculations, which are often evident only on prolonged observation. As the disease progresses, muscle atrophy spreads proximally. Spasticity gradually appears as well; it is usually only mild at first and may indeed remain so over the ensuing course of the disease. The intrinsic muscle reflexes are usually brisk, more than one would expect in view of the concomitant atrophy and weakness, but py-
Fig. 7.14 Spinal muscular atrophy in a 46-year-old woman. There is marked atrophy of the muscles of the shoulder girdles, arms, and hands, as well as of the paravertebral musculature.
Fig. 7.15 Atrophy of the tongue due to true bulbar palsy in a 65- year-old woman with amyotrophic lateral sclerosis.
ramidal tract signs are not necessarily demonstrable. The bulbar muscles are also involved in about 20 % of patients, as manifested by atrophy, weakness, and fasciculations of the tongue (Fig. 7.15), dysarthria, and dysphagia (true bulbar palsy). Involvement of the corticobulbar tracts is indicated by exaggerated nasopalpebral, perioral and masseteric reflexes, and by involuntary laughter and crying, which are often present.
Treatment. Riluzole marginally slows the progression of the disease. There is no other specific treatment.
Prognosis. ALS takes a chronically progressive course. Death usually ensues within one or two years, although a minority of patients survives longer.
7
Diseases of the Spinal Cord
aövnqaop
Mumenthaler / Mattle, Fundamentals of Neurology © 2006 Thieme akblpk jqß
All rights reserved. Usage subject to terms and conditions of license.