


83
6Diseases of the Brain and Meninges
Congenital and Perinatally Acquired Diseases |
Infectious Diseases of the Brain and Meninges . . . |
111 |
|||||
of the Brain . . . |
83 |
|
|
Metabolic Disorders and Systemic Illnesses |
|
||
Traumatic Brain Injury . . . |
87 |
|
Affecting the Nervous System |
. . . 120 |
|
||
Intracranial Pressure and Brain Tumors . . . |
92 |
Diseases of the Basal Ganglia . . . |
127 |
|
|||
Circulatory Disorders of the Brain and |
|
Cerebellar Diseases . . . |
135 |
|
|
||
Nontraumatic Intracranial Hemorrhage |
. . . 98 |
Dementing Diseases . . |
. 137 |
|
|
||
Congenital and Perinatally Acquired Diseases of the Brain
Fundamentals
|
The developing brain is vulnerable to damage by a |
|
Disturbances of movement of many different |
|
|
number of different pathological mechanisms. The |
|
kinds; the more common ones are summarized |
|
|
variably severe neurological deficits that result are |
|
in Table 6.1. These are usually accompanied by a |
|
|
known collectively as cerebral movement dis- |
|
variably severe delay of motor development. |
|
|
orders or infantile cerebral palsy (CP). In general, |
|
Mental retardation (sometimes designated |
|
|
this term implies the presence of deficits of multi- |
|
“childhood psycho-organic syndrome”) is com- |
|
|
ple types: |
|
mon and is characterized by the delayed acqui- |
|
|
|
|
|
|
|
|
|
|
|
Table 6.1 The most important cerebral movement disorders
Name of disorder |
Clinical features |
Pathoanatomical substrate |
Causes |
|
|
|
|
infantile spastic |
spasticity, predominantly in the legs; |
pachymicrogyria (abnormally |
perinatal injury (disturbance |
diplegia |
pes equinus, scissor gait, mentally |
hard, small gyri) |
of cerebral development, |
(Little disease) |
often normal |
|
embryopathy, severe neonatal |
|
|
|
jaundice) |
congenital cerebral |
usually, paresis of arm and face |
porencephaly (cavities in the |
birth trauma (asphyxia, |
monoparesis |
|
brain parenchyma), localized |
hemorrhage) |
|
|
atrophy |
|
congenital |
arms more severely affected than |
porencephaly |
birth trauma (asphyxia, |
hemiparesis |
legs, seizures in ca. 50 %, usually |
|
hemorrhage) |
|
mentally impaired |
|
|
congenital quadri- |
arms more severely affected than |
porencephaly, bilateral; |
birth trauma (asphyxia, |
paresis (bilateral |
legs, occasionally bulbar signs, |
often hydrocephalus |
hemorrhage), also prenatal |
hemiparesis) |
seizures; severe mental impairment |
|
injury |
congenital pseudo- |
dysphagia to liquids, dysarthria, |
bilateral lesions of the |
prenatal injury or birth |
bulbar palsy |
usually not mentally impaired |
corticobulbar pathways |
trauma, congenital malforma- |
|
|
|
tion (syringobulbia) |
atonic−astatic |
generalized flaccid weakness, inabil- |
frontal lobe atrophy cerebellar |
|
syndrome (Foerster) |
ity to stand, impaired coordination, |
defects |
|
|
severe mental impairment |
|
|
bilateral athetosis |
athetotic or other involuntary move- |
basal ganglionic defects, status |
disturbances of cerebral |
(athétose double) and |
ments, often combined with spastic |
marmoratus (multiple confluent |
development, perinatal injury, |
congenital chorea |
paresis |
gliotic areas in the basal ganglia); |
esp. severe neonatal jaundice |
(choreoathetosis) |
|
status dysmyelinisatus (Vogt) in |
|
|
|
cases of later onset |
|
congenital rigor |
rigor without involuntary move- |
status marmoratus |
disturbances of cerebral |
|
ments, postural abnormalities, no |
|
development, perinatal injury, |
|
pyramidal tract signs, severe mental |
|
esp. severe neonatal jaundice |
|
impairment, seizures |
|
|
congenital cerebellar |
gait ataxia, intention tremor and |
cerebellar developmental |
disturbances of cerebellar |
ataxia |
impaired coordination, motor |
anomalies |
development |
|
developmental retardation, dys- |
|
|
|
arthria, possibly in combination with |
|
|
|
other motor syndromes |
|
|
|
|
|
|
blubber blubber
Mumenthaler / Mattle, Fundamentals of Neurology © 2006 Thieme All rights reserved. Usage subject to terms and conditions of license.
6
Diseases of the Brain and Meninges
84 6 Diseases of the Brain and Meninges
|
|
seizures), and certain symptoms may worsen over the |
sition of mental abilities, by impaired attention, |
|
|
|
course of the individual’s life. |
|
and often also by hyperactivity and inability to |
|
|
|
|
|
concentrate. The term psychomotor retardation |
|
|
refers to a combination of movement distur- |
|
Special Clinical Forms |
bances and mental retardation. |
|
|
|
|
|
Epileptic seizures often arise later on. |
|
Some of the more important etiological types of early |
|
|
|
Common types and their causes. Tables 6.1, 6.2 pro- |
childhood brain damage will now be discussed in- |
|
dividually: |
||
vide an overview of the major types of brain damage |
|
|
that are present at birth, or acquired in early childhood, |
Hydrocephalus is a pathological dilatation of the inner |
|
and their causes. These include genetic disorders, cere- |
(and sometimes also the outer) cerebrospinal fluid |
|
bral hypoxia during the birth process, birth trauma, in- |
spaces. Various types of hydrocephalus are listed in |
|
trauterine infections (rubella embryopathy, toxoplas- |
Table 6.3. In terms of pathogenesis, the most common |
|
mosis, cytomegalovirus, syphilis, HIV), and chronic in- |
type of hydrocephalus in childhood is occlusive hydro- |
|
toxications (alcohol embryopathy). Prematurity and dif- |
cephalus (gliosis, stenosis, or malformation of the aque- |
|
ficult delivery are the most important risk factors. |
|
|
Possible indications of brain damage in the newborn |
|
|
include cyanosis at birth, a weak cry, and hypotonia. In |
|
|
the early prenatal period, there may be further abnor- |
|
|
malities of muscle tone, as well as pathological reflexes |
|
|
(cf. p. 43 ff.). Later on, a squint or left-handedness may |
|
|
be a sign of brain damage. |
|
|
Treatment consists of physical therapy (e. g., of the |
|
|
Bobath or Vojta type), which should be begun as early as |
|
|
possible, making use of the child’s reflex behavior, as |
|
|
well as special education and rehabilitation. The goal of |
|
|
treatment is maximal independence. |
|
|
Prognosis. Although the neurological deficits of cere- |
|
|
bral palsy do not progress over time, certain manifesta- |
|
|
tions may not appear until later in life (e. g., epileptic |
|
|
Table 6.2 Important causes of congenital and perinatally acquired brain damage
Perinatal asphyxia
(Genetically determined) structural anomalies of the brain, e.g.:
microcephaly
meningoencephalocele
meningomyelocele
micropolygyria
Arnold−Chiari malformation, with or without hydrocephalus
Phakomatoses
tuberous sclerosis (Bourneville disease)
encephalofacial angiomatosis (Sturge−Weber disease)
neurofibromatosis (von Recklinghausen disease)
von Hippel−Lindau disease
Brain damage acquired in utero
rubella embryopathy
congenital toxoplasmosis
congenital cytomegaly
congenital syphilis
congenital HIV infection
alcohol embryopathy
Severe neonatal jaundice (due to Rh incompatibility)
Synostosis and craniostenosis
Traumatic intracranial hemorrhage during delivery
subdural hematoma
intracerebral hemorrhage
intraventricular hemorrhage
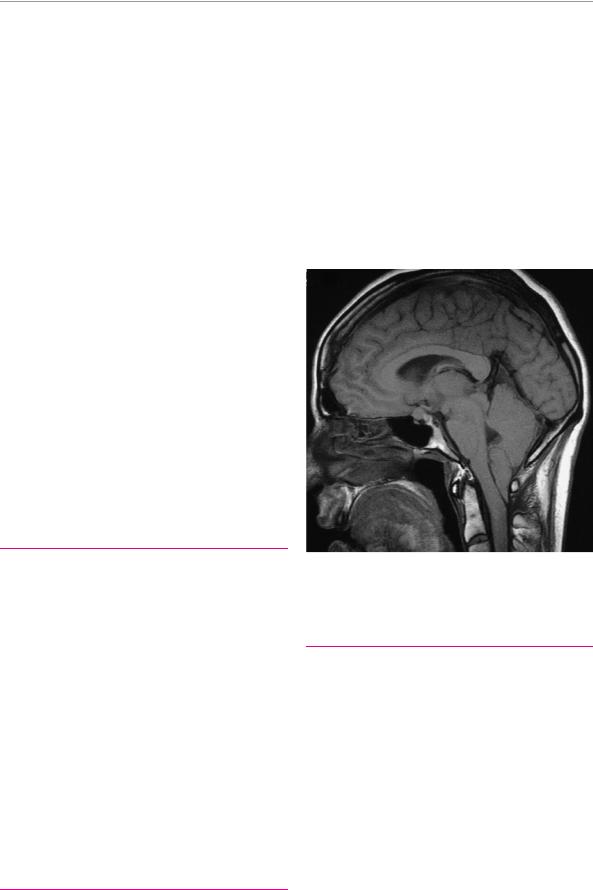
Fig. 6.1 Arnold−Chiari malformation (MR image). The cerebellar tonsils are caudally displaced below the arch of the atlas deep into the cervical spinal canal (courtesy of Dr. D. Huber, Radiological Institute, Hirslanden−Klinik, Zurich).
Table 6.3 Types and terminology of hydrocephalus
Internal |
enlargement of the ventricles: |
hydrocephalus |
|
obstructive |
due to obstruction of CSF flow within the |
|
ventricular system (e. g., aqueductal stenosis) |
|
or at its exits (e. g., obstruction of foramina |
communicat- |
of Magendie and Luschka) |
nonobstructive internal hydrocephalus |
|
ing |
|
malabsorptive |
a subtype of communicating hydrocephalus |
|
due to impaired CSF resorption (e. g., cister- |
|
nal adhesions or dysfunction of the pacchion- |
|
ian granulations) |
External |
enlargement of the subarachnoid space over |
hydrocephalus |
the cerebral convexities and/or in the cisterns |
External and |
combination of the above |
internal |
|
hydrocephalus |
|
Hydrocephalus |
internal and external hydrocephalus second- |
ex vacuo |
ary to brain atrophy |
|
|
Mumenthaler / Mattle, Fundamentals of Neurology © 2006 Thieme All rights reserved. Usage subject to terms and conditions of license.

Congenital and Perinatally Acquired Diseases of the Brain
duct, Arnold−Chiari malformation [Fig. 6.1] with impaired outflow through the foramina of Luschka and Magendie). In the Arnold−Chiari malformation, part of the medulla and the cerebellar tonsils are displaced below the foramen magnum into the cervical spinal canal. This anomaly may be combined with internal hydrocephalus and syringomyelia.
There may also be communicating hydrocephalus, whose etiology and pathogenesis are often unclear.
The chief clinical sign of hydrocephalus in childhood is abnormal enlargement of the head, which may already be noted in a prenatal ultrasound study or at birth, and which progresses over time. Protrusion of the frontal bone and depression of the orbital plate make the upper part of the sclera visible and cause the lower part of the iris to sink below the lower lid, in the so-called “settingsun sign.” The essential diagnostic tests are CT and/or MRI. The treatment, if required, is neurosurgical, usually the implantation of a ventriculoperitoneal or ventriculoatrial shunt. The prognosis of isolated hydrocephalus, in the absence of other neurological abnormalities, is good: once the hydrocephalus is treated, two-thirds of children go on to have a normal physical and mental development.
Microcephaly is usually due to prenatal toxic influences
(e. g., alcohol) or infections (e. g., cytomegalovirus), or to genetic factors. Affected persons are generally of lower than normal intelligence.
Dysraphic malformations. The most common type is spina bifida with meningomyelocele: in this disorder, there is a closure defect of the posterior arches of multiple vertebrae, usually in the lumbosacral region, accompanied by a prolapse of the meninges and spinal cord through the defect. The level and extent of spinal cord involvement determine whether paralysis of the lower limbs will be manifest at birth. Even if the defect is surgically repaired in the first few hours after birth, major sensorimotor impairment and urinary disturbances generally persist. This type of malformation may be accompanied by internal hydrocephalus and by anomalies of the craniocervical junction, which often require treatment. Other dysraphic syndromes include acrania (partial or total absence of the skull), anencephaly (absence or degeneration of most of the brain, with acrania— practically always a fatal condition), and encephalocele (prolapse of the meninges and brain tissue through a defect in the bony skull).
Areas of neuronal heterotopia (i. e., islands of gray matter anomalously lying outside the cerebral cortex) may be found in the periventricular zones or in a subcortical layer (lamina) that creates the appearance of a “double cortex” on MRI. Subcortical laminar heterotopia
(SCLH) is a genetic disorder of dominant inheritance caused by a mutation of the doublecortin gene on the X chromosome. The disorder is more severe, and often lethal, in males, because they lack a normal copy of the gene. In surviving males, SCLH is often combined with lissencephaly (“smooth brain,” i. e., absence of the cerebral gyri and sulci). Heterotopia is a common cause of epilepsy.
Ulegyria is a type of early childhood brain damage characterized by scarring and abnormally small gyri (microgyria). These structural abnormalities can be seen on MRI.
Phakomatoses are genetic disorders that cause complex malformations and tumors predominantly affecting the ectodermally derived structures of the body, i. e., the brain, peripheral nervous system, and skin. They are also called neurocutaneous disorders. The internal organs may also be affected. An overview is provided in Table 6.4.
The main types of brain disorder acquired in utero are the following:
Rubella embryopathy occurs in 10 % of infants that have been exposed by maternal infection in the first trimester of pregnancy. The associated anomalies include cataracts, deafness, microcephaly, and heart defects.
Congenital toxoplasmosis occurs when the fetus is infected in the second half of gestation by maternal infection in a mother without previous exposure to Toxoplasma gondii. Its manifestations include psychomotor retardation, convulsions, progressive hydrocephalus, and visual disturbances due to chorioretinitis. Plain radiographs and CT reveal intracerebral calcifications.
Congenital cytomegalovirus infection causes premature birth and low birth weight, microcephaly, hydrocephalus, convulsions, periventricular calcifications, and abnormalities in organs outside the nervous system as well.
Congenital HIV infection occurs in one-quarter of all babies born to HIV-positive mothers. It causes encephalopathy with psychomotor retardation, as well as immune deficiency, with later complications.
Congenital syphilis is now rare. Its typical stigmata include a saddle nose, cutaneous fissures at the corners of the mouth, and later crescentic defects of the teeth (Hutchinson teeth), interstitial keratitis, and hearing loss.
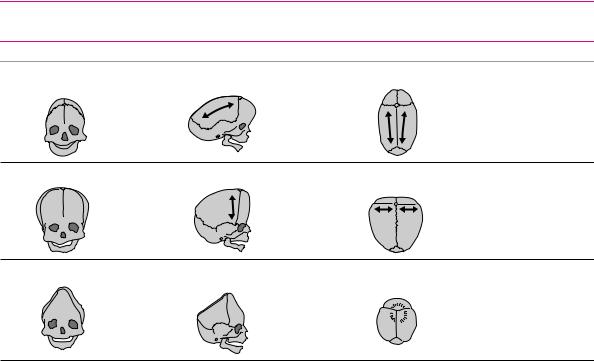
Malformations of the skull come in many different forms: there are dysraphic malformations characterized by faulty closure of the cranial vault (cranioschisis), premature closure of the cranial sutures (craniostenosis), and anomalies of the craniocervical junction including basilar impression, platybasia, Arnold−Chiari syndrome (p. 84), and Dandy−Walker syndrome (malformation of the posterior fossa with aplasia of the inferior portion of the cerebellar vermis, cystic enlargement of the fourth ventricle, and occlusive hydrocephalus). The more common types of craniosynostosis are listed and illustrated in Table 6.5.
blubber blubber
Mumenthaler / Mattle, Fundamentals of Neurology © 2006 Thieme All rights reserved. Usage subject to terms and conditions of license.
85
6
Diseases of the Brain and Meninges
86 |
6 Diseases of the Brain and Meninges |
|
|
||
|
|
|
|
|
|
|
|
Table 6.4 The most important phakomatoses |
|
|
|
|
|
|
|
|
|
|
|
Disease |
Neuropathology and |
Further features |
Age of onset and other |
|
|
|
clinical neurological manifestations |
|
remarks |
|
|
|
|
|
|
|
|
tuberous sclerosis |
glial tumors (giant cell astrocy- |
multiple (fibro-)adenomas in the |
salaam seizures often appear |
|
|
(Bourneville disease) |
tomas, “tubers”) in the ventricular |
face (typically butterfly-shaped, |
as early as infancy; autosomal |
|
|
|
walls and surface of the brain, often |
adenoma sebaceum) as well as |
dominant inheritance |
|
|
|
calcified; as a result, thickening and |
on the gums and nails; adenomas |
|
|
|
|
sclerosis of the gyri of the cerebral |
in the heart, kidney, and retina |
|
|
|
|
and cerebellar cortex. Clinical |
|
|
manifestations: mental retardation, epileptic seizures
encephalofacial |
calcified, mixed capillary and venous |
angiomatosis |
angioma of the leptomeninges, |
(Sturge−Weber |
usually unilateral, with reactive atro- |
disease) |
phy and gliosis of the neighboring |
|
brain parenchyma; serpentine in- |
|
tracranial calcification is a typical |
|
radiograph finding. Possible clinical |
|
manifestations: epileptic seizures, |
|
mental retardation, and sometimes |
|
hemiparesis |
choroidal hemangioma and angioma of the face (nevus flammeus) on the same side as the intracranial lesion
onset in early childhood; sporadic or dominant inheritance pattern, with variable penetrance
von Hippel−Lindau |
cystic hemangioblastoma, usually in |
disease |
a cerebellar hemisphere, causing pro- |
|
gressive cerebellar signs and signs of |
|
intracranial hypertension |
neurofibromatosis |
multiple neurofibromas of periph- |
(von Recklinghausen |
eral nerves, nerve roots (particularly |
disease) |
in the cauda equina) and cranial |
|
nerves; |
|
intracranially, in some cases, bi- |
|
lateral acoustic neuroma and/or |
|
meningioma (in neurofibromatosis |
|
type II), or optic glioma and/or |
|
astrocytoma (type I); clinically, pro- |
|
gressive radicular or peripheral |
|
nerve deficits (flaccid paresis, |
|
sensory deficits), signs and symp- |
|
toms of a cerebellopontine angle |
|
tumor (esp. hearing loss, tinnitus), |
|
visual impairment with optic glioma |
retinal angiomatosis; less commonly, cystic changes of other internal organs (esp. kidney, pancreas, epididymis)
variable number and density of nodular skin lesions, which may be either broad-based or pedunculated (cutaneous neurofibroma); café-au-lait spots
onset in middle age; autosomal dominant inheritance
the cutaneous changes are often present at birth or become apparent in early childhood; they typically become very prominent in puberty. Malignant degeneration is possible. Autosomal dominant inheritance, with frequent new mutations
Table 6.5 Types of craniosynostosis
Type |
Fused suture |
Shape of head |
Remarks |
scaphocephaly |
sagittal suture |
long, narrow (“boat-shaped”) |
|
(=dolichocephaly) |
|
|
|
|
|
|
most common form |
acrocephaly |
coronal suture |
high, broad on top; |
|
flat forehead |
|
||
|
|
|
oxycephaly |
sagittal, coronal, |
pointed |
and lambdoid sutures |
|
|
|
|
second most common form
brachycephaly coronal and lambdoid sutures short, broad
|
|
|
|
Mumenthaler / Mattle, Fundamentals of Neurology © 2006 Thieme |
Continued |
||
All rights reserved. Usage subject to terms and conditions of license. |
|||
|
|||