

Биомедицина практика
.pdfму для обнаружения однонуклеотидных замен применяют и другие подходы. В одном из них объединены ПЦР и метод, основанный на лигировании олигонуклеотидных зондов (ЛОЗ), – ПЦР/ЛОЗ.
Предположим, что в определенном сайте нормального гена (скажем, в 106-м положении) находится пара А-Т, а в том же сайте мутантного гена – G-C. Зная нуклеотидные последовательности, фланкирующие 106-й нуклеотид, можно синтезировать два коротких (20-нуклеотидных) фрагмента, прилегающих к данному сайту и комплементарных противоположным цепям. Основная особенность этой пары олигонуклеотидов состоит в том, что 3'-концевой нуклеотид одного из них (зонд X) комплементарен основанию, находящемуся в 106-м положении нормальной последовательности, а 5'- концевой нуклеотид второго (зонд Y) комплементарен нуклеотиду, примыкающему к 106-му нуклеотиду. При отжиге этих зондов с содержащей нормальную последовательность ДНК-мишенью (амплифицированной методом ПЦР) происходит их полная гибридизация, и при добавлении в реакционную смесь ДНК-лигазы зонды Х и Y ковалентно сшиваются. Если же эти зонды отжигаются с мутантной ДНК, в которой произошла замена 106-го нуклеотида, то некомплементарный ему 3'-концевой нуклеотид зонда Х не может образовать с ним пару. И хотя зонд Y по-прежнему гибридизуется полностью, ДНК-лигаза не может сшить зонды Х и Y. Можно синтезировать и другие олигонуклеотидные зонды, полностью соответствующие последовательности с мутантным 106-м нуклеотидом. При таком наборе зондов лигирование будет происходить в случае их отжига с мутантной ДНКмишенью и не будет в случае отжига с нормальной мишенью. Таким образом, метод ПЦР/ЛОЗ различает две ситуации: лигирование зондов и отсутствие его.
Чтобы определить, произошло ли лигирование, 5'-конец зонда Х метят биотином, а 3'-конец зонда Y – дигоксигенином, низкомолекулярным соединением, связывающимся с соответствующим антителом. После гибридизации и лигирования проводят денатурацию ДНК для высвобождения гибридизовавшегося зонда и переносят смесь в пластиковую лунку, покрытую стрептавидином. Лунку промывают, чтобы удалить весь материал, кроме связавшегося со стрептавидином биотинилированного зонда. Затем добавляют в лунку антитела к дигоксигенину, предварительно соединенные со щелочной фосфатазой. После промывания (удаления несвязанного конъюгата) добавляют бесцветный хромогенный субстрат. Окрашивание раствора в лунке свидетельствует о связывании антитела к дигоксигенину с зондом, меченным дигоксигенином, т.е. о том, что этот зонд был лигирован с зондом, меченным биотином. Если же окрашивания не происходит, значит, лигирования не было.
Располагая двумя парами зондов, можно установить генетический статус любого человека. Например, ДНК гетерозиготных носителей дает поло-
41
жительный ответ с обеими парами зондов; ДНК лиц, обладающих двумя копиями нормального гена, – только с тем набором зондов, который содержит нуклеотид, комплементарный нормальному сайту; и, наконец, ДНК индивидов с двумя измененными копиями гена – только с набором зондов, детектирующим мутантный сайт. Чтобы минимизировать необходимое для анализа количество исходной ДНК, перед гибридизацией участок ДНК-мишени, содержащий тестируемый сайт, амплифицируют с помощью ПЦР. ПЦР/ЛОЗ является быстрым, чувствительным и высокоспецифичным методом. Все стадии роботизированы, что позволяет проводить до 1200 тестов в день.
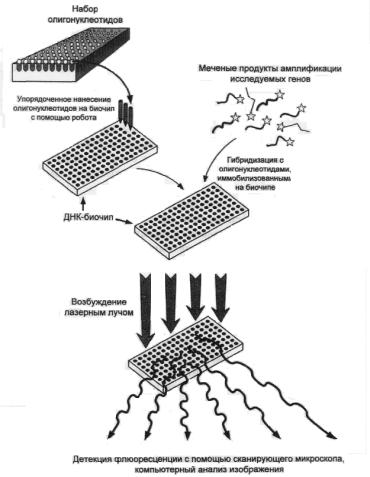
Использование ДНК-биочипов
Этот метод является одним из наиболее перспективных в современной молекулярной биологии (рис. 9). ДНК-микрочип (олигонуклеотидный микрочип) представляет собой миниатюрную пластину-матрицу, в ячейки которой с помощью специальных автоматизированных микрометодов нанесен упорядоченный набор из десятков, сотен и даже тысяч иммобилизованных олигонуклеотидов.
Рис. 9. Технология ДНК-биочипов (общий принцип)
42
Данные олигонуклеотиды специфически гибридизуются с разнообразными флюорохромно-меченными молекулами ДНК (например, продуктами амплификации исследуемых генов или молекулами кДНК – продуктами обратной транскрипции тканевых РНК), после чего образовавшиеся дуплексы облучаются лазером, а результат флюоресценции фиксируется с помощью сканирующего конфокального микроскопа и анализируется с использованием соответствующего компьютерного программного обеспечения.
В формате биочипов могут быть реализованы многие из таких методов анализа нуклеотидных последовательностей, как автоматическое секвенирование, «минисеквенирование» и др. Последние достижения в данной области связаны с применением фотолитографических и полупроводниковых технологий, позволяющих упорядоченно фиксировать на микрочипе до 400 000 и более олигонуклеотидов (каждый – на участке площадью около 20 микрон!), а также с разработкой методов синтеза олигонуклеотидов in situ, основанных на доставке необходимых реагентов в нужные сайты микрочипа с помощью устройств, действующих по принципу струйного принтера.
Применение на практике технологии ДНК-биочипов принципиально позволяет проводить автоматизированный и эффективный анализ большого числа известных мутаций и полиморфизмов, осуществлять быстрое секвенирование крупных фрагментов ДНК, а также одномоментно анализировать экспрессию большого числа генов изучаемой ткани в заданный интервал времени, что сулит поистине уникальные возможности в изучении основ функционирования генома.
43
БИОИНФОРМАТИКА
Под биоинформатикой обычно понимают использование компьютеров для решения биологических задач.
Биоинформатика оформилась как самостоятельная дисциплина в начале 80-х годов XX века. Поводом к возникновению этой области науки послужило открытие эффективного метода определения нуклеотидных последовательностей. В1978–80 гг. были определены несколько полных последовательностей размером около 4 тыс. оснований (букв) – плазмиды pBR322, вируса саркомы зеленой мартышки SV40, бактериофага ФХ-174. Стало ясно, что последовательности длиной в несколько тыс. оснований не могут быть проанализированы вручную. Появилась необходимость в компьютерных методах обработки таких массивов информации. Задачи, которые тогда стояли часто были очень простыми – найти открытую рамку, сайты рестрикции, сходство двух последовательностей, оттранслировать нуклеотидную последовательность в белок. Этот круг задач определялся потребностями экспериментаторов. Хотя уже в то время стали формулироваться и более сложные задачи, некоторые из них до сих пор нельзя считать окончательно решенными, – поиск генов, физическое картирование, поиск сигналов в последовательностях, предсказание вторичной структуры РНК. Биоинформатика оформилась как самостоятельная, хотя и вспомогательная, ветвь науки в январе 1980 г., когда вышел специальный компьютерный выпуск биологического журнала «Nucleic acids research».
Впрочем, первые работы по биоинформатике, посвященные анализу последовательностей, появились раньше (в конце 60-х – начале 70-х годов ХХ века). Это теперь уже классические работы по сравнению аминокислотных последовательностей белков – построение матрицы сравнения аминокислотных остатков (Дейхофф), алгоритма выравнивания последовательностей (Нидельман, Вунш). Другое направление исследований было связано с анализом возможных вторичных структур транспортных РНК. В результате Эйген обнаружил, что все тРНК можно уложить в характерную структуру, похожую на клеверный лист. Этот результат сразу вошел в учебники по молекулярной биологии, и только через 12 лет было экспериментально показано, что это предсказание верно.
Следующий этап связан с увеличением количества прочитанной информации. Количество расшифрованных последовательностей перевалило за тысячу, размеры последовательностей стали достигать сотни тысяч. Это привело к тому, что возникла необходимость хранения и доступа к информации. Так появились первые банки последовательностей – банк ЛосАламос, база данных EMBL, базы данных по аминокислотным последовательностям. Появился новый класс задач – быстрый поиск сходства в банках последовательностей. Следует отметить, что объем прочитанной ин-
44
формации примерно совпадал со скоростью роста мощности компьютеров удвоение за полтора года. Однако некоторые задачи имеют нелинейную зависимость сложности от размера данных. Поэтому по-прежнему остро стоят вопросы создания эффективных алгоритмов.
Ситуация стала меняться с появлением проектов расшифровки полных геномов, в том числе генома человека. Если до этого периода задачи ставились так – есть последовательность, про которую уже многое известно; найти что-нибудь еще. Теперь же компьютерный анализ выходил на первый план. Требовалось проанализировать полный геном и найти в нем новые особенности – новые гены, сигналы регуляции и т.п. И если раньше компьютерный анализ шел после эксперимента, то теперь результаты компьютерного анализа часто определяют направление исследований. Более того, часто данные компьютерного анализа являются самостоятельными биологически значимыми результатами.
В настоящее время биоинформатика стала существенным элементом биологической науки, способной не только обслуживать потребности экспериментаторов, но также и продуцировать собственные результаты.
Биоинформатика в подавляющем большинстве мировых научных центров понимается как синоним вычислительной молекулярной биологии.
Есть несколько основных направлений этого раздела науки, в зависимости от исследуемых объектов:
•биоинформатика последовательностей;
•структурная биоинформатика;
•компьютерная геномика.
Цели и задачи биоинформатики
Основополагающий принцип биоинформатики состоит в том, что биополимеры, например, молекул нуклеиновых кислот и белков могут быть преобразованы в последовательности цифровых символов. Кроме того, для представления мономеров аминокислотных и нуклеотидных цепей необходимо лишь ограниченное число алфавитных знаков.
Подобная гибкость анализа биомолекул с помощью ограниченных алфавитов привела к успешному становлению биоинформатики. Развитие и функциональная мощь биоинформатики во многом зависят от прогресса в области разработки аппаратных средств и программного обеспечения ЭВМ. Простейшие задачи, стоящие перед биоинформатикой, касаются создания и ведения баз данных биологической информации.
По сути, предмет биоинформатики включает в себя три компонента:
1)создание баз данных, позволяющих осуществлять хранение крупных наборов биологических данных и управление ими;
2)разработка алгоритмов и методов статистического анализа для определения отношений между элементами крупных наборов данных;
45
3) использование этих средств для анализа и интерпретации биологических данных различного типа – в частности, последовательностей ДНК, РНК и белков, белковых структур, профилей экспрессии генов и биохимических путей.
Цели
Цели биоинформатики следующие:
1)Организовывать данные таким образом, чтобы исследователи имели доступ к текущей информации, хранящейся в базах данных, и могли вносить в нее новые записи по мере получения новых сведений.
2)Развивать программные средства и информационные ресурсы, которые помогают в управлении данными и в их анализе.
3)Применять эти средства для анализа данных и интерпретации полученных результатов таким образом, чтобы они имели биологический смысл.
Задачи
В целом задачи биоинформатики состоят в анализе информации, закодированной в биологических последовательностях. Последнее предполагает следующее:
1.Обнаружение генов в последовательностях ДНК различных орга-
низмов.
2.Развитие методов изучения структуры и (или) функции новых расшифрованных последовательностей и соответствующих структурных областей РНК.
3.Определение семейств родственных последовательностей и построение моделей.
4.Выравнивание подобных последовательностей и восстановление филогенетических деревьев с целью выявления эволюционных связей.
Помимо перечисленных выше задач, следует упомянуть еще один важнейший вопрос биоинформатики – обнаружение мишеней для медикаментозного воздействия и отыскание перспективных опытных соединений.
Предмет
Биоинформатика осуществляет следующие виды деятельности.
1)Управление биологическими данными и их обработка; сюда входит их организация, отслеживание, защита, анализ и т. д.
2)Организация связи между учеными, проектами и учреждениями, вовлеченными в фундаментальные и прикладные биологические исследования. Связь может включать в себя электронную почту, пересылку файлов, дистанционный вход в систему, телеконференции, электронные информационные табло и, наконец, учреждение сетевых информационных ресурсов.
46
Организация наборов биологической информации, документов и литературы, а также обеспечение доступа к ним, их поиска и выборки.
3) Анализ и интерпретация биологических данных с применением вычислительных методов, как-то: визуализация, математическое моделирование, а также построение алгоритмов высокопараллельной обработки сложных биологических структур.
Прикладная область биоинформатики
В расшифровке смыслового содержания последовательностей наметились два различных аналитических направления:
1)согласно первому подходу ученые опираются на методы распознавания регулярных комбинаций, посредством которых обнаруживают подобие последовательностей и, следовательно, выявляют эволюционно связанные структуры и функции;
2)согласно второму подходу используют методы предсказания ah initio – для прогнозирования трехмерных структур и, в конечном счете, выведения функции непосредственно по линейной последовательности. Прямое предсказание трехмерной структуры белка по его линейной последовательности аминокислот – важнейшая цель биоинформатики.
Анализ гомологичности последовательностей
Одна из движущих сил биоинформатики – поиск подобий между разными биомолекулами. Помимо систематической организации данных, идентификация белковых гомологов имеет прямое практическое применение. Теоретические модели белков обычно основаны на структурах близких гомологов, определенных опытным путем.
Всякий раз, когда ощущается недостаток биохимических или структурных данных, могут быть выполнены исследования на дрожжеподобных низших организмах, а результаты могут быть распространены на гомологичные молекулы более высоких организмов, например, человека. Более того, данный подход упрощает проблему понимания сложных геномов: за счет непосредственного анализа простых организмов и последующего распространения тех же самых принципов на более сложные. Это могло бы привести к опознаванию потенциальных мишеней для медикаментозного воздействия путем испытаний на гомологах основных микробных белков.
Разработка лекарственных препаратов
Опирающийся на биоинформатику подход к открытию лекарств дает важное преимущество. С помощью биоинформатики могут быть описаны генотипы, сопряженные с патофизиологическими состояниями, что в принципе позволит опознать соответствующие молекулярные мишени. Посредством программного транслятора по известной последовательности нуклео-
47
тидов может быть определена вероятная аминокислотная последовательность кодируемого белка.
В случае принятия такого подхода методы изучения последовательностей могли бы применяться для поиска гомологов у опытных организмов; и на основании подобия последовательностей было бы возможно моделировать структуру конкретного белка, взяв за основу экспериментально установленные структуры. И наконец, стыковочные алгоритмы могли бы проектировать молекулы, потенциально связывающиеся с моделируемой структурой, прокладывая тем самым путь для биохимических испытаний, проверяющих биологическую активность этих молекул уже на физическом белке.
Прогнозирующие функции
Благодаря технологии массового просмотра данных можно получить ответ на ряд вопросов, касающихся эволюционных, биохимических и биофизических характеристик исследуемых биомолекул. Возможно установить: а) специфические мотивы в структуре белков, соответствующие определенным филогенетическим группам, б) общность между различными мотивами, наблюдаемыми у отдельных организмов, в) долю аналогичных мотивов, общих для родственных организмов, г) степень родства, выведенного из тривиальных эволюционных деревьев, и д) различие метаболических путей у разных организмов.
Кроме того, на основании того факта, что мотивы в структуре белка часто связаны с определенными биохимическими функциями, можно получать данные относительно функций белка. На основании анализа структурных данных можно составить карту взаимодействий всех белков того или иного организма.
Применение в медицине
Приложения к медицинским наукам затрагивают главным образом анализ экспрессии генов. Как правило, это предполагает сбор данных об экспрессии в клетках, пораженных различными заболеваниями, и сравнение этих измерений с нормальными уровнями экспрессии. Обнаружение генов с измененной в пораженных клетках экспрессией создает основу для объяснения причин болезни и указывает потенциальные мишени для лекарственных препаратов.
Располагая подобной информацией, можно разрабатывать соединения, которые связываются с экспрессируемым белком. Далее могут быть проведены эксперименты на микроматрицах, чтобы оценить реакцию на фармакологическое воздействие полученного опытного соединения; подобная информация может помочь также в разработке тестов для обнаружения или прогноза токсичности опытных лекарств на стадии клинических испытаний.
Объединение биоинформатики с экспериментальной геномикой может открыть путь для многих достижений, которые приведут к коренным
48
изменениям в будущих программах здравоохранения. К ним можно отнести послеродовое определение генотипа с целью оценки восприимчивости или устойчивости индивидуума к определенным болезням и патогенам, предписание уникального сочетания вакцин, уменьшение затрат на лечение за счет повышения эффективности терапии и предупреждения рецидивов заболевания. Вкупе все эти новшества могут привести к разработке индивидуальных пищевых рационов и выявлению заболеваний на ранних стадиях.
Кроме того, программы медикаментозного лечения могли бы специально подбираться к конкретному пациенту и болезни, и таким образом обеспечивать наиболее эффективный курс лечения с минимальными побочными эффектами. В частности, проект «Геном человека» принес несомненную пользу судебной медицине и фармацевтической промышленности, привел к открытию многих полезных и вредных генов, внес неоценимый вклад в развитие представлений об эволюции человека и, кроме того, способствовал разработке методов диагностики болезней, возможных осложнений и генетически обусловленных реакций на терапевтическое воздействие, а также развитию индивидуальных подходов к лечению, методов обнаружения мишеней для лекарственных препаратов и, наконец, становлению генотерапии.
Биоинформатика последовательностей
Этот раздел биоинформатики занимается анализом нуклеотидных и белковых последовательностей. В настоящее время разработаны эффективные экспериментальные методы определения нуклеотидных последовательностей. Определение нуклеотидных последовательностей стало рутинной хорошо автоматизированной процедурой.
В результате рутинной хорошо автоматизированной процедуры уже получено огромное количество генетических текстов. Так, в базе данных EMBL на 15.02.2007 хранится 87 000 493 документов с описанием нуклеотидных последовательностей, содержащих в целом 157 545 686 001 символов (нуклеотидов), что соответствует примерно библиотеке в 105 толстых томов с убористым шрифтом. Найти нужный ген в EMBL, это все равно, что найти цитату в такой библиотеке. Без помощи компьютера сделать это очень трудно. А число данных экспоненциально растет.
Например, геном небольшой бактерии – это непрерывная строка длиной в 1–10 миллионов символов, и далеко не вся ДНК кодирует белки. Первый тип биоинформатической задачи – это задача поиска в нуклеотидных последовательностях особых участков: кодирующих белки, кодирующих РНК (например, тРНК), локусов связывания с регуляторными белками и других. И это не всегда простые задачи, например, гены эукариотических организмов состоят из чередующихся «осмысленных» и «бессмысленных» фрагментов (экзонов и интронов), и расстояние между «осмысленными»
49
фрагментами может достигать тысяч нуклеотидов. После того, как найден ген, необходимо выявить, что он кодирует и какую функциональную нагрузку несет.
Если речь идет об участке ДНК, кодирующем белок, то с помощью весьма простой операции – трансляции с использованием известного генетического кода можно получить соответствующие аминокислотные последовательности. Из известных на сегодня 4 273 512 белков около 94 % последовательностей – это именно такие гипотетические трансляты, и больше о них ничего не известно.
Но биологические объекты – это объекты, возникшие в процессе эволюции. Сравнительно-эволюционный подход – один из мощнейших подходов в биологии. Например, функция белка из одного организма хорошо экспериментально изучена, в другом организме нашли белок с похожей аминокислотной последовательностью. Можно предположить, что второй (неизвестный) белок выполняет ту же или схожую функцию. И здесь сразу возникает несколько вопросов. Во-первых, что значит похожая последовательность? Как сравнивать последовательности? При какой степени сходства последовательностей можно предполагать, что белки выполняют сходные функции?
Сравнение последовательностей (выравнивание) является важнейшей задачей биоинформатики.
Крупным успехом биоинформатики является создание программы Blastp и ClustalX. Но в настоящее время постоянно совершенствуют методы выравниваний.
Можно привести много примеров того, как сравнительноэволюционный подход в сочетании с биоинформатическими методами порождает новое биологическое знание.
Генетические тексты – тексты с большой долей шума, сравнивая родственные последовательности, в ряде случаев удается отфильтровать шум и выявить сигнал, например, короткую последовательность нуклеотидов, способную связываться с белком-регулятором, или аминокислотные остатки в ферменте, отвечающие за связывание субстрата. Чтобы быть уверенными в результате, в биоинформатике используют теорию вероятности и математическую статистику.
Таким образом, основные задачи биоинформатики, связанные с анализом отдельных последовательностей, состоят в следующем:
1.Выравнивание и определение сходства двух последовательностей.
2.Построение множественных выравниваний.
3.Распознавание генов.
4.Предсказание сайтов связывания регуляторных белков.
5.Предсказание вторичной структуры РНК.
50