

Биомедицина практика
.pdfплификационных тест-систем позволяет определять несколько сот копий в исследуемом образце.
4.Актуальность ответа (быстрота получения результата). Высокая технологичность и автоматизация метода позволяет получить результаты исследования в руки врача и пациента в день проведения анализа.
5.Возможность доклинической и ретроспективной диагностики.
ПЦР позволяет осуществить определение патогена или дефектного гена в организме еще до развития заболевания. Например, при инфекциях в инкубационном периоде, т.е. серонегативной фазе или при латентном характере заболевания. Кроме того, возможно проведение ПЦР в архивном (фиксированном) материале или биологических остатках, что важно для идентификации личности или отцовства.
6.Проведение анализа возможно в минимальном объеме пробы
(до нескольких микролитров), что крайне важно в неонатологии, судебной медицине, клинической генетике и т.п.
7.Возможность одновременной диагностики нескольких возбуди-
телей заболеваний или аномальных генов в одной пробе без ущерба для чувствительности или специфичности результата.
8.Возможность экспертизы. Полученные результаты ПЦР возможно вносить в компьютерные информационные носители или фотографии для оценки независимыми экспертами.
Несмотря на вышеуказанные достоинства, метод ПЦР все же не лишен некоторых недостатков, которые следует учитывать при оценке результатов исследований. В настоящее время выделяют следующие две группы.
Первая – технологическая, подразумевающая под собой высочайшие требования к оснащению лаборатории, качеству тест-наборов и строжайшее соблюдение регламента исследования во избежание получения ложных результатов. Решение проблемы качества анализов возможно при соответствующей квалификации персонала и обязательной сертификации лаборатории. В связи с этим врачи должны требовать от лаборатории, проводящей исследования методом ПЦР, государственный сертификат.
Вторая – клиническая, заключающаяся в неоднозначной прогностической оценке положительного результата ПЦР. Именно этот факт зачастую является необоснованным аргументом практических врачей для сомнения в полученном результате и эффективности метода ПЦР. Поясним на примере. Показано, что только у 40–60 % лиц с обнаруженной в крови методом ПЦР ДНК цитомегаловируса клинически может развиться заболевание. В данной ситуации при оценке результатов исследования совершается типичная ошибка, заключающаяся в отождествлении двух принципиально различных понятий – «инфицированность» и «инфекционная болезнь».
Таким образом, недостатки ПЦР лежат не в сути метода, а в неправильном методическом подходе (алгоритме лабораторной диагностики) при
31
обследовании пациента и неверной клинической интерпретации полученных результатов.
Возможности применения метода ПЦР в лабораторной диагностике
Наиболее эффективно и обоснованно использование метода ПЦР
ДНК:
–в урологической и гинекологической практике – для выявления хламидиоза, уреаплазмоза, гонореи, герпеса, гарднереллеза, микоплазменной инфекции, ВПЧ – вируса папилломы человека;
–в пульмонологии – для дифферециальной диагностики вирусных и бактериальных пневмоний, туберкулеза;
–в гастроэнтерологии – для выявления геликобактериоза;
–в клинике инфекционных заболеваний – в качестве экспресс-метода диагностики сальмонеллеза, дифтерии, вирусных гепатитов B, C, G.
–в гематологии – для выявления цитомегаловирусной инфекции, онковирусов, ВИЧ.
Лигазная цепная реакция
Метод впервые описан в 1989 г. ЛЦР – это метод амплификации, включающий последовательные циклы лигирования (соединения) четырех олигонуклеотидных праймеров, комплементарных цепям ДНК-матрицы. В нем используется способность ДНК-лигазы соединять 2 пары комплементарных олигонуклеотидов после их гибридизации с последовательностями мишени in vitro.
Основными компонентами ЛЦР являются: лигаза, 2 или 4 специфических праймера и буфер для цепной реакции.
Линейная амплификация специфических праймеров происходит при лигировании одной пары олигонуклеотидов. При использовании двух пар олигонуклеотидов, одна из которых комплементарна верхней цепи ДНКматрицы, а вторая – нижней, достигается экспоненциальная амплификация. На первом этапе происходит гибридизация двух пар олигонуклеотидных праймеров с ДНК-матрицей при температуре +65 °С. Затем происходит лигирование олигонуклеотидов в месте их соединения с образованием более длинного продукта, связанного с матрицей. Специфичность реакции обеспечивается тем, что любое нарушение гомологии в области стыка двух олигонуклеотидов сразу же предотвращает их лигирование. На втором этапе реакционную смесь нагревают до +94 °С. При этом происходит денатурация и отделение лигированного продукта от матрицы. При последующем охлаждении до +65 °С процессы гибридизации и лигирования повторяются. Вновь образованные продукты лигирования выступают в качестве матриц в после-
32
дующих циклах реакции. В ходе ЛЦР происходит экспоненциальное увеличение количества исходных продуктов лигирования. Выявление продуктов амплификации можно проводить с использованием ферментных меток.
Молекулярная диагностика генных болезней
К настоящему времени на хромосомах человека картировано около 1500 генов, мутации которых приводят к различным наследственным заболеваниям. Молекулярная клиническая диагностика генных заболеваний дает ответ на вопрос, входят ли обследуемые индивидуумы или их потомки в группу повышенного генетического риска. ДНК-анализ можно использовать для выявления носителей генов наследственных заболеваний, а также для пренатальной и пресимптоматической диагностики генетических нарушений. Раньше применялись биохимические методы, основанные на выявлении продукта анализируемого гена. Решающим преимуществом молекулярной диагностики является то, что объектом исследования является непосредственно молекула ДНК больного. ДНК-тесты не требуют экспрессии мутантного гена для его выявления, что позволяет разработать системы скрининга для всех заболеваний. Для молекулярной клинической диагностики генных болезней можно использовать, например, методы блотгибридизации по Саузерну, ПЦР и другие.
Методы первичной идентификации мутаций
Наиболее точная информация о природе мутаций может быть получена с помощью метода ДНК-секвенирования, позволяющего определить первичную последовательность нуклеотидов в определенном фрагменте ДНК. Однако, ввиду сравнительно крупных размеров генов и отсутствия автоматических ДНК-секвенаторов во многих диагностических лабораториях, к методу секвенирования обычно приступают уже после того, как с помощью других методов обнаружено наличие мутации в определенном фрагменте гена, и остается только выяснить ее нуклеотидную природу. Поэтому сканирование мутаций обычно начинают с использования методических приемов, позволяющих улавливать нуклеотидные замены сразу в достаточно протяженных участках ДНК. К таким приемам относят: метод анализа конформационного полиморфизма однонитевой ДНК (SSCP), метод электрофореза в денатурирующем геле (DGGE), метод химического расщепления некомплементарных сайтов (СМС), метод гетеродуплексного анализа (НА).
Секвенирование ДНК
Существует 2 основных метода секвенирования: химический – метод Максама–Гильберта и дидезоксисеквенирование – метод Сэнджера. В первом случае используют химическое расщепление ДНК по одному основа-
33

нию, во втором – синтезируют нужную цепь ДНК in vitro, специфически останавливая синтез на заданном основании (рис. 7).
Чаще при секвенировании используют метод Сэнджера, так как он более надежен и прост в исполнении. На первом этапе ДНК денатурируют, чтобы получить однонитевые молекулы. Затем добавляют секвенирующий праймер – искусственно синтезированную олигонуклеотидную последовательность, комплементарную определенному участку исходной молекулы ДНК. Создают условия для гибридизации праймера, то есть для образования двухцепочечного участка, и инициируют синтез ДНК, добавляя в реакционную смесь ДНК-полимеразу и дезоксинуклеотидтрифосфаты – dATP, dCTP, dGTP и dTTР, один из которых является радиоактивным. Синтез ведут в четырех параллельных пробирках, в каждую из которых добавляют один из специфических дидезоксинуклеотидтрифосфатов, или терминато-
ров синтеза – ddATP, ddCTP, ddGTP или ddTTР (ddNTP).
Рис. 7. Секвенирование ДНК по методу Сэнджера
34
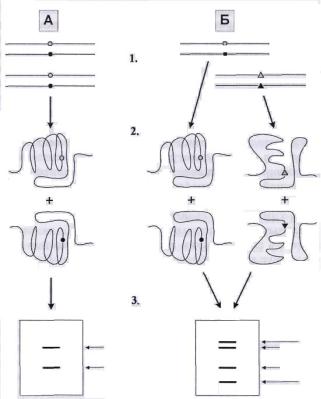
При встраивании ddNTP на место соответствующего нуклеотида синтез ДНК прекращается. Таким образом, в каждой из пробирок получают набор различающихся по длине радиоактивно меченных фрагментов ДНК с одним и тем же специфическим для данной пробирки дидезокситерминатором на конце молекулы. После одновременного электрофоретического разделения этих фрагментов на четырех соседних дорожках и авторадиографии размер синтезированных фрагментов может быть определен, а значит, и определена локализация дидезоксинуклеотидов и порядок соответствующих им нуклеотидов в исходной молекуле ДНК. На каждом секвенирующем геле может быть определена первичная последовательность всего около 500 пар оснований.
Метод анализа конформационного полиморфизма однонитевой ДНК (Single Strand Conformation Polymorphism – SSCP)
Метод предложен M. Orita в 1989 г., основан на регистрации различий в электрофоретической подвижности одинаковых по величине, но различающихся по пространственной организации (вследствие нуклеотидных замен) однонитевых фрагментов ДНК.
Рис. 8. SSCP-анализ (общий принцип)
Скручивание (конформация) небольших однонитевых участков ДНК существенно зависит от их нуклеотидной последовательности, так что за-
35
мена даже одного основания в молекулах одинакового размера приводит к изменению их пространственной структуры (рис. 8). Метод включает амплификацию специфических сегментов ДНК размером от 50 до 300 п.о., обычно в присутствии меченых динуклеотидтрифосфатов, денатурацию образовавшихся продуктов ПЦР и нативный высокоразрешающий ЭФ в ПААГ. Иногда амплификацию проводят без использования метки, но тогда для выявления ДНК на электрофореграммах используют более чувствительные по сравнению с окраской бромидом этидия методы, например, окраску азотнокислым серебром. На конформацию оказывают влияние различные внешние факторы – температура, концентрация акриламида и глицерина в геле, ионная сила буферных растворов. Оптимальный подбор этих параметров позволяет эффективно разделять амплифицированные фрагменты ДНК, различающиеся по длине всего на один нуклеотид.
Метод денатурирующего градиентного гель-электрофореза
(Denaturation Gradient Gel Electrophoresis – DGGE)
Метод основан на различиях электрофоретической подвижности нормальных и мутантных амплифицированных двухнитевых фрагментов ДНК при ЭФ в денатурирующем геле. Денатурация может происходить за счет разницы температур, различной концентрации мочевины или формальдегида. При этих условиях одинаковые по величине двухнитевые молекулы ДНК, отличающиеся по нуклеотидной последовательности, денатурируют по-разному. Объясняется это тем, что G-C связь более прочна по сравнению со связью между нуклеотидами А-Т. Кроме того, наличие участка негомологичного спаривания значительно облегчает денатурацию ДНК. Разработан компьютерный алгоритм, позволяющий предсказывать характер плавления в зависимости от нуклеотидной последовательности ДНК. При электрофорезе амплифицированных двухнитевых фрагментов ДНК в геле с линейно возрастающим градиентом концентрации денатурирующих агентов плавление нитей ДНК происходит в строго специфичной для данной последовательности области, эквивалентной температуре плавления – Тm, т.е. такой температуре, при которой каждая пара оснований с 50%-й вероятностью может соединиться или разойтись. После начала плавления ДНК продвижение ее фрагментов в геле резко замедляется до тех пор, пока не наступит полная денатурация молекул. В результате будет происходить разделение фрагментов ДНК, различающихся по нуклеотидному составу. Метод очень чувствителен и, в отличие от SSCP, применим для более крупных амплифицированных фрагментов ДНК. Метод может быть с успехом применен для анализа индуцированных мутаций, т.к. позволяет улавливать мутации даже в 1 из 100 клеток, обработанных мутагеном.
К недостаткам метода следует отнести технические сложности, связанные с трудностями получения хорошего градиента денатурирующего
36
агента в ПААГ. Это требует специального технического оснащения. Другим недостатком метода является сложность детекции мутаций, расположенных на концах амплифицированных фрагментов. Связано это с тем, что при прохождении ДНК через гель может начаться частичная денатурация концов молекул еще до достижения оптимальной области плавления. Поэтому мутации, локализованные недалеко от границ амплифицированного участка ДНК, оказывают меньшее влияние на процесс плавления и за счет этого могут не выявляться. Во избежание этого к концам исследуемой амплифицированной геномной ДНК пришивают GC-последовательности размером в несколько десятков пар оснований. Такие монотонные тугоплавкие участки выполняют роль своеобразных зажимов на концах молекул, и шансы обнаружения мутаций выравниваются, независимо от их локализации внутри амплифицированного фрагмента.
Метод гетеродуплексного анализа (Heteroduplex analysis – HA)
Принцип метода – возникновение (образование) гетеродуплексов между нормальной и несущей точковую мутацию цепочками ДНК в образце гетерозиготной по данной мутации особи. Такие гетеродуплексные двухцепочечные ДНК благодаря конформационным особенностям в местах несовпадения нуклеотидов имеют иную электрофоретическую подвижность, чем соответствующие гомодуплексы. Хотя эти различия могут быть обнаружены при электрофорезе в обычном ПААГ, значительно более эффективное разделение гомо– и гетеродуплексов может быть достигнуто при использовании новых вариантов гелей – Hydrolink либо MDE. Детекция мутаций осуществляется как изотопным, так и неизотопными методами.
Метод химического расщепления мест (нуклеотидного)
несоответствия (Сhemical Mismatch Cleavage – СМС)
Метод основан на свойствах некоторых химических агентов специфически разрывать нить ДНК в месте нарушения гомологичного спаривания в результате несоответствия пар оснований вследствие точечных мутаций, т.е. в местах возникновения гетеродуплексов. С этой целью амплифицированную нормальную последовательность ДНК смешивают с избытком аналогичной тестируемой ДНК или РНК, проводят денатурацию (отжиг), после чего нормальная и тестируемая ДНК ренатурирует с образованием гетеродуплексов, которые подвергаются химическому расщеплению. Цитозин чувствителен к действию гидроксиламина, а тимин – к тетраоксиду осмия. Последующая обработка пиперидином приводит к полному разрыву молекулы ДНК в модифицированном сайте. Выявление мутаций осуществляют с помощью меченых ДНК-зондов, соответствующих, как правило, нормальной последовательности ДНК. Такими зондами могут служить синтезированные олигонуклеотиды, клонированные последовательности ДНК или амплифицированные
37
фрагменты. После обработки соответствующими химическим агентами идентификация и локализация мутантных сайтов в тестируемом фрагменте ДНК проводится с помощью электрофореза. Современные модификации метода позволяют идентифицировать до 95–100 % мутаций.
Большими преимуществами этого метода являются: 1) возможность исследовать протяженные участки ДНК – до 2 тыс. п.о., 2) способность одновременно выявить и локализовать несколько мутаций в тестируемом фрагменте ДНК и 3) возможность одновременного использования нескольких ДНК-зондов для поиска мутаций – мультиплексный вариант методики.
К числу недостатков можно отнести высокую токсичность используемых химических реактивов. Последняя может быть частично ослаблена использованием карбодимида для идентификации GT-гетеродуплексов.
Все рассмотренные выше методы детекции мутаций предполагают в качестве обязательного последующего этапа секвенирование содержащих изменения сегментов ДНК с целью точной идентификации нуклеотидных замен, оценки их фенотипического проявления и определения причастности к развитию болезни. Поэтому рассмотренные методы редко используются в практической диагностике и при популяционном скрининге гетерозигот.
Молекулярное сканирование известных мутаций
Детекцию уже известных мутаций можно проводить более простыми способами, не требующими последующего секвенирования.
Амплификация – рестрикция
Мутации, изменяющие длину амплифицированных фрагментов, могут быть выявлены с помощью нативного ЭФ в ПАА или агарозном гелях.
Наиболее просто диагностируются те замены нуклеотидов, которые приводят к исчезновению или образованию сайта узнавания для какой-нибудь из рестриктаз, что легко выявляется по изменению длины амплифицированного фрагмента ДНК после его обработки соответствующей эндонуклеазой. Поэтому сразу после идентификации мутации проводится компьютерный поиск возможных сайтов рестрикции в месте локализации замены основания.
Расщепление амплифицированных ДНК-фрагментов проводится по прописям, рекомендованным фирмой-изготовителем рестриктаз, в буфере, прилагаемом к ферменту.
Данный метод применяют, например, для диагностики серповидноклеточной анемии.
Серповидноклеточная анемия – это генетическое заболевание, обусловленное заменой одного из нуклеотидов в кодоне, который соответствует шестой аминокислоте в β-цепи молекулы гемоглобина. У индивидов, гомозиготных по мутантному гену (S/S), эритроциты имеют необычную серповидную форму; это связано с искажение конформации молекулы гемо-
38
глобина вследствие замены в ней валина на глутаминовую кислоту. Мутантный гемоглобин не может с достаточной эффективностью переносить кислород, и у таких больных развивается тяжелая анемия с прогрессирующим поражением сердца, легких, мозга, суставов и других органов. У индивидов, гетерозиготных по данному гену (A/S) (носителей генетического заболевания), эритроциты имеют нормальную форму, и симптомы заболевания проявляются лишь в экстремальных условиях (на большой высоте над уровнем моря либо при слишком высоких или низких температурах, когда снижается снабжение организма кислородом). Если оба родителя гетерозиготны (имеют генотип A/S), то вероятность того, что их ребенок будет гомозиготным по мутантному гену (S/S) (т. е. будет болен серповидноклеточной анемией), составляет 25 %.
Замена одного нуклеотида в β-глобиновом гене, приводящая к серповидноклеточной анемии, сопровождается элиминацией сайта для рестрицирующей эндонуклеазы Cvn1. Этот фермент узнает последовательность CCTNAGG и расщепляет молекулу ДНК между основаниями С и Т (N – любой из четырех нуклеотидов). В нормальном гене эта последовательность имеет вид CCTGAGG, а в гене серповидноклеточной анемии – ССТGTGG. На этом различии основывается ДНК-диагностика данного заболевания. Используя праймеры, фланкирующие сайт Cvn1, амплифицируют с помощью ПЦР небольшое количество тестируемой ДНК. Амплифицированный фрагмент обрабатывают Cvn1, продукты рестрикции разделяют с помощью гель-электрофореза и окрашивают их бромистым этидием. При наличии Cvn1-сайта на электрофореграмме появляется специфический набор полос, отличный от такового в отсутствие Cvn1-сайта. Описанным способом можно быстро установить генетический статус обследуемого, не проводя при этом процедуру гибридизации.
ПЦР-опосредованный сайт-направленный мутагенез
Если естественных рестрикционных сайтов в месте мутации найти не удается, то такие сайты могут быть созданы искусственно. В частности, разработана методика создания с помощью ПЦР новых сайтов рестрикции в мутантныхаллелях– методПЦР-опосредованногосайт-направленногомутагенеза.
Для этого амплифицируемый участок ДНК выбирают таким образом, чтобы 3’-конец одного из праймеров непосредственно примыкал к мутантному сайту. Именно этот праймер не полностью комплементарен матричной ДНК. В нем изменяют один из нуклеотидов с 3’-конца так, чтобы в сочетании с нуклеотидом мутантного, но не нормального сайта в этом месте образовывался сайт рестрикции для какой-нибудь из известных нуклеаз. Тогда после рестрикции и электрофореза продуктов амплификации геномной ДНК у индивидуумов, не содержащих данную мутацию, на электрофореграмме будет присутствовать один нерестрицированный фрагмент, у ге-
39
терозигот появятся два дополнительных фрагмента, соответствующим по длине рестрицированным сегментам ДНК, и у гомозигот по мутации будут присутствовать только эти два рестрицированных фрагмента.
Обнаружение мутаций в разных сайтах одного гена
Далеко не все генетические заболевания обусловливаются одним изменением в гене. В большинстве случаев мутации могут возникать в разных сайтах в пределах одного гена, но приводят к одному генетическому заболеванию. В качестве примера можно привести β-талассемию – наследственное заболевание, связанное с утратой активности β-глобина. У гетерозиготных носителей при этом наблюдается небольшая анемия. Индивиды же, гомозиготные по одному из как минимум восьми возможных мутантных сайтов, для поддержания жизни нуждаются в регулярном переливании крови и другом лечении. Поскольку мутация в любом из восьми специфических сайтов β- глобинового гена может приводить к β-талассемии, необходимо провести по крайней мере восемь разных тестов. Такая диагностика возможна, хотя дорогостоящая. Поэтому для скрининга мутаций в разных сайтах одного гена была разработана стратегия ПЦР/гибридизация, основанная на проведении одной реакции. Для этого синтезируют набор специфических 20-нуклеотидных зондов, каждый из которых полностью комплементарен фрагменту генамишени, несущему известную мутацию. К 3'-концу каждого присоединен гомополимер poly(dT) длиной примерно 400 нуклеотидов, с помощью которого ДНК-зонд связывается с заранее отмеченной точкой на найлоновом фильтре, а остальная его часть остается свободной и может гибридизоваться. Сегменты тестируемой ДНК, каждый из которых включает по одному из возможных мутационных сайтов, одновременно амплифицируют с помощью ПЦР, причем один праймер из каждой пары на 5'-конце помечен биотином. Амплифицированные фрагменты ДНK-мишени гибридизуют с зондами, пришитыми к фильтру, в условиях, обеспечивающих гибридизацию только полностью комплементарных последовательностей. В гибридизационную смесь добавляют стрептавидин, связанный с щелочной фосфатазой (можно использовать пероксидазу хрена или уреазу). После гибридизации промывают фильтр и добавляют неокрашенный субстрат. Если имеет место полное соответствие между амплифицированным сегментом ДНК-мишени и специфическим зондом, то на фильтре появится цветная точка. На один и тот же фильтр можно нанести несколько точек, соответствующих целому ряду разных специфических зондов. Проанализировав эту цветную мозаику, можно идентифицировать один из возможных сайтов мутации.
Метод ПЦР/ЛОЗ
Не все генетические нарушения, приводящие к появлению дефектных генов, сопровождаются утратой или изменением сайтов рестрикции, поэто-
40