

Lectures.Part.2
.2.pdf51
і залежить від властивостей і складу системи. (Товщина дифузійного шару λ приймається такою, що потенціал адсорбційного шару на відстані δ-λ зменшується в е разів).
В щільній частині ПЕШ іони утримуються не лише за рахунок електростатичних сил, а й за рахунок специфічної адсорбції, тобто не кулонівськими силами. В розчинах, що містять поверхнево-активні іони, їх кількість в адсорбційному шарі може бути такою, що їх заряд перевищить заряд поверхні.
Потенціал в адсорбційному шарі при збільшенні відстані від поверхні зменшується до потенціалу дифузійного шару лінійно, а далі за експоненційним законом.
Електрокінетичний потенціал дана теорія прив’язує до площини ковзання між твердою і рідкою фазою, при їх відносному русі.
Рис.9.3. а)Розподіл зарядів відносно поверхні; б) зміна потенціалу відносно поверхні; в) зміна концентрації іонів відносно поверхні за теорією Штерна
δ – товщина адсорбційного шару λ – товщина дифузійного шару ПК - площина ковзання φ0 – потенціал поверхні
φδ – потенціал адсорбційного шару ζ – електрокінетичний потенціал
Якщо протиіони здатні то специфічної адсорбції то в щільній частині ПЕШ їх може адсорбуватись стільки, що сумарний заряд перевищить заряд поверхні. Тобто, потенціали φ0
Рис.9.4. Залежність потенціалу від відстані в умовах специфічної адсорбції протиіонів за теорією Штерна
та φδ матимуть різний знак. Це явище називають перезарядка поверхні.
© Ю.Павловський 2011
52
Специфічна адсорбція залежить від спорідненості іонів, що адсорбуються до поверхні, їх здат-
ності утворювати недисоційовані поверхневі речовини. Великий адсорбційний потенціал мають багатовалентні іони Al+3, Fe+3, Ti+3, Th+4, Ce+4, PO4-3… та органічні іони.
Будова міцели
В дисперсних системах подвійний електричний шар виникає на поверхні частинок. Частинка дисперсної фази разом з іонами подвійного електричного шару, що на ній утворився називається міцелою.
Розглянемо будову міцели, що утворюється внаслідок наступної реакції:
FeSO4 + 2NaOH = Fe(OH)2 (тв.) + Na2SO4
Утворення колоїдного розчину при протіканні хімічної реакції можливо лише у тому випадку, коли хоча б один з продуктів реакції утворює нову фазу.
У даному випадку гідроксид заліза (II) нерозчинний у воді і утворить агрегати з m молекул
Fe(OH)2.
Поверхня цих новоутворених агрегатів має дуже велику надлишкову енергію і на ній з розчину будуть адсорбуватись іони добудовуючи кристалічну ґратку. Цей процес підпорядковується правилу вибіркової адсорбції іонів Панета-Фаянса: на твердій поверхні переважно адсорбуються ті іони, що мають однакове з поверхнею атомне угрупування, або ізоморфні до поверхні. Іони, які добудовують кристалічну ґратку носять назву «потенціалотвірні».
Агрегат разом з шаром «потенціалтвірних» іонів складають ядро міцели.
На поверхні ядра з розчину адсорбується іони протилежного до заряду ядра знаку, утворюючи адсорбційний шар «протиіонів». Ядро разом з адсорбційним шаром «протиіонів» складає заряджену частинку.
Інша частина «протиіонів» знаходиться у розчині поблизу частинки і складає дифузійний шар «протиіонів». Частинка з дифузійним шаром «протиіонів» і є міцелою.
Будова міцели залежить від того який з електролітів взятий у надлишку. Якщо у надлишку FeSO4 , то іони Fe2+ є «потенціалтвірними», а SO42– – «протиіонами»; якщо у надлишку NaOH, то іони OH– будуть «потенціалтвірними», а Na+ – «протиіонами».
а) будова міцели при надлишку речовини А (FeSO4): [mFe(OH)2] – агрегат міцели
[mFe(OH)2]∙nFe+2 – ядро міцели
{[mFe(OH)2]∙nFe+2(n-x) SO4-2 }+2x– частинка міцели (зарядження позитивно)
{[mFe(OH)2]∙nFe+2(n-x) SO4-2 }+2x ∙xSO4-2 – власне міцела;
b) будова міцели при надлишку речовини В (NaOH):
{[mFe(OH)2]∙nOH–(n-x) Na+ }–x ∙x Na+
частинка міцели зарядження негативно.
Вплив різних факторів на ПЕШ
1.Додавання індиферентних електролітів Неіндиферентними називають електроліти, які містять іони, здатні добудовувати
кристалічну ґратку та утворювати нерозчинні сполуки з потенціалтвірними іонами. Усі решта – індиферентні.
а) При додаванні до колоїдного розчину індиферентних електролітів, що мають у своєму складі ті ж протиіони, що є в адсорбційному шарі, дифузійний шар стискається. Дифузний потенціал зменшується. Потенціал поверхні не змінюється.
При певній концентрації електроліту дифузний шар може стиснутись до моноіонного і перетворитись в шар Гельмгольца; ζ-потенціал зменшиться до нуля. Такай стан дисперсної системи називають ізоелектричним;
©Ю.Павловський 2011
53
б) При додаванні електроліту, що не має спільного протиіона , може відбуватись обмін іонів згідно з їх положенням в ліотропних рядах; в) Якщо валентність нового протиіона більша, то рівновага буде сильно збільшена в бік іона більшої валентності;
г) При додаванні електроліту, що містить високовалентні іони, здатні до специфічної адсорбції можлива перезарядка поверхні (див. Рис.9.4).
2.Вплив неіндиферентних електролітів а) При додаванні електроліту, що містить іон здатний добудовувати кристалічну ґра-
тку, потенціал поверхні φ0, потенціал адсорбційного шару φδ та електрокінетичний ζ- потенціал зростають.
б) Якщо електроліт містить іони, які утворюють з потенціалтвірними нерозчинну
сполуку, то потенціал поверхні потенціал поверхні φ0, потенціал адсорбційного шару φδ та електрокінетичний ζ-потенціал змінюють свій знак на протилежний.
3.Розведення розчину приводить до збільшення товщини λ-шару і ζ-потенціалу. Сильне розведення до десорбції потенціалтвірних іонів і зменшення потенціалу поверхні φ0.
4.Підвищення температури викликає дію аналогічну розведенню.
5.Природа дисперсійного середовища, також впливає на ПЕШ: чим більша полярність молекул середовища, тим більший ζ-потенціал.
Електрокінетичні явища
При накладанні електричного поля на дисперсну систему її частинки починають переміщатись до одного з електродів. Це явище називають електрофорезом. Якщо дисперсна фаза не може рухатись (пориста мембрана) то рухається дисперсійне середовище, в системі спостерігається електроосмос.
Електрофорез та електроосмос свідчать про те, що на поверхні дисперсної фази існує подвійний електричний шар: поверхня і сусідні шари середовище заряджені протилежно.
Основними факторами, від яких залежить швидкість електрофорезу та електроосмосу є електрокінетичний або ζ-потенціал, напруженість електричного поля, в’язкість середовища. Електрокінетичний потенціал виникає на межі ковзання дисперсної фази та середовища, при їх русі одне відносно іншого і є однозначною характеристикою електричних властивостей дисперсної фази яка, крім того, піддається вимірюванню.
Для знаходження ζ-потенціалу вимірюють швидкість руху частинок при електрофорезі Uеф або електроосмосі Uео. За рівнянням Гельмгольца-Смолуховського:
Uеф |
|
0 |
H |
(9.2) |
|
|
|
|
|||
|
|
|
|
|
|
Uео |
|
|
0 |
H |
(9.3) |
|
|
|
|||
|
|
|
|
|
|
Розрахунок ζ-потенціалу за рівняннями (9.2-9.3) деколи дає занижені результати, що пояснюється ефектом релаксації та електрофоретичним гальмуванням.
Ефект релаксації – зміна сферичної симетрії дифузійного шару навколо частинки в результаті руху фаз у різних напрямках. Це приводить до поляризації частинок, виникнення їх власного поля, яке направлене протилежно прикладеному, як наслідок, зменшення швидкості їх руху.
Електрофоретичне гальмування – зустрічний рух протиіонів, що створює додаткове тертя, яке протидіє рухові частинок.
В багатьох випадках експериментальні факти не вкладаються в теоретичні рамки оскільки швидкість руху частинок залежить ще й від розмірів частинок дисперсної фази та температури.
© Ю.Павловський 2011
54
Крім приведених вище, є ще два електрокінетичні явища. Це, так званий, ефект Дорна або «потенціал протікання» і «потенціал осідання».
Ефект Дорна полягає у виникненні потенціалу на електродах, які поміщені по обидва боки мембрани, через яку під тиском протікає дисперсійне середовище (явище обернене до електроосмосу).
Потенціал осідання виникає на електродах розміщених на різних рівнях в посудині з частинками, що осідають. Це явище обернене до електрофорезу.
© Ю.Павловський 2011

55
Лекція 10. Коагуляція і стабілізація дисперсних систем
Коагуляцією називають процес зменшення агрегативної стійкості колоїдної системи, що приводить до укрупнення частинок за рахунок їх злипання і руйнування колоїдної системи. Розрізняють дві стадії коагуляції – приховану і явну.
Перша, прихована стадія проходить дуже швидко. На цій стадії частинки хоч і збільшуються, але осад не утворюється. Можливі зовнішні зміни: помутніння, поява зависі, зміна забарвлення тощо.
Друга, явна стадія наступає в результаті подальшої агрегації частинок, яка за деякий час завершується повним розділенням системи на дві фази і випаданням дисперсної фази в осад – коагулят або коагель, що має певну структуру.
Коагуляцію колоїдних систем викликають:
1)додавання електролітів;
2)змішування протилежно заряджених золів;
3)зміна складу дисперсійного середовища;
4)зміна температури, кип’ятіння, заморожування;
5)механічна дія: перемішування або струшування;
6)вібрація та ультразвук;
7)пропускання електричного струму;
8)опромінення світлом та елементарними частинками.
Дія усіх факторів зводиться до одного – вони якимось чином руйнують енергетичний бар’єр і метастабільна система переходить у більш стійкий стан в результаті коагуляції.
Найбільш важливе значення і найбільш вивченим є процес коагуляції дисперсних систем електролітами.
Основне поняття в теорії коагуляції золів електролітами – поріг коагуляції: мінімальна концентрація електроліту в об’ємі системи, в якій спостерігається коагуляція даного золю:
ce Ve , моль/м3 (10.1)
Vз Ve
Величина обернена до порогу коагуляції називається коагулююча здатність. Ця величина показує, який об’єм золю може коагулювати, при додаванні 1 моля електроліту.
Є кілька теорій коагуляції золів електролітами.
За адсорбційною теорією коагуляції Фрейндліха, при коагуляції золів іони коагулятори адсорбуються частинками дисперсної фази. Процес підпорядковується рівнянню ізотерми Фрейндлі-
ха (7.3).
Втрата стійкості в даній теорії пояснюється зменшенням заряду частинки. Багатовалентні іони, які проявляють більшу адсорбційну здатність, коагулюють золі в менших концентраціях ніж одновалентні.
Для підтвердження теорії проводилась коагуляція золю Fe(OH)3 розчинами Na2HP*O4 і K2S*O4 з радіоактивними ізотопи фосфору та сірки. Коагулят був радіоактивним, а отже адсорбував аніони.
Електрична теорія коагуляції базується на тому, що при додаванні електроліту змінюється подвійний електричний шар. Заряд поверхні частинки φ0 не змінюється, але в результаті стискання дифузійного шару ζ-потенціал падає до певного критичного значення. Стабільність золю зменшується і він коагулює. Мінімальний ζ-потенціал при якому спостерігається коагуляція називають критичним потенціалом золю.
© Ю.Павловський 2011

56
Теорія стійкості і коагуляції дисперсних систем ДЛФО (Дерягіна-Ландау-Фервея- Овербека)
За цією теорією стійкість дисперсної системи визначається балансом сил притягання і відштовхування, які виникають між частинками, що знаходяться у неперервному боунівському русі при їх зближенні.
Енергія взаємодії частинок складається з енергії електростатичного відштовхування і енергії міжмолекулярного притягання Ван-дер-Ваальса:
U = UE + UM. (10.2)
Позитивна енергія електростатичного відштовхування зростає при зближенні частинок за експоненційним законом, а негативна енергія притягання за степеневим:
UE e h (10.3)
U 1 (10.4)
M |
h 2 |
|
На малих відстанях, h < 2÷10Å, переважає притягання і тому на потенціальній кривій взаємодії знаходиться перший мінімум (І), що відповідає злипанню частинок. Глибина цього мінімуму UI
= (20÷25)∙kT (рис.10.1).
Рис. 10.1.Криві електростатичного відштовхування (UE), міжмолекулярного притягання (UM) та сумарна потенціальна крива взаємодії (U)
На великих відстанях, h = 100÷1000Å, також переважає притягання, оскільки степенева функція спадає повільніше ніж експонента. Мінімум ІІ відповідає притяганню частинок через шар середовища. Глибина цього мінімуму UII = −(0÷10)∙k∙T.
На середніх відстанях, 10 < h < 100Å, переважає відштовхування. Максимум ІІІ характеризує потенціальний бар’єр, що перешкоджає злипанню частинок. Глибина бар’єру UIII = (0÷100)∙kT і залежи від концентрації електроліту в системі.
За видом сумарної кривої можна зробити висновок про стійкість дисперсної системи. Розрізняють три характерних види потенціальних кривих (рис.10.2).
Крива 1 відповідає стану системи з високим потенціальним бар’єром і відсутністю вторинного мінімуму. Система є агрегативно стійкою.
Крива 2 вказує на наявність досить високого енергетичного бар’єру і невеликого мінімуму. Частинки мають змогу об’єднуватись у флокули на відстанях, що відповідають другому мінімуму. Частинки у флокулах безпосередньо не контактують, бо розділені шаром середовища. Наявність бар’єру перешкоджає їх подальшому зближенню. Цей стан відповідає зворотності коагуляції. При зниженні енергетичного бар’єру можлива коагуляція, при усуненні вторинного мінімуму – пептизація.
© Ю.Павловський 2011

57
Рис. 10.2. Характерні криві потенціальної взаємодії між частинками
Крива 3 відповідає такому стану дисперсної систем, коли при будь-якій відстані між частинками переважає притягання. В системі спостерігається швидка коагуляція.
В теорії ДЛФО розрізняють два граничні випадки коагуляції: нейтралізаційну коагуляцію та концентраційну коагуляцію.
Нейтралізаційна коагуляція спостерігається в системах в яких потенціал поверхні φ0 знижений в результаті нестачі потенціалтвірних іонів або внаслідок специфічної адсорбції протилежно заряджених іонів. При зниженні заряду електричні сили відштовхування слабшають, енергетичний бар’єр знижується і частинки мають змогу злипатись. Концентрації електроліту, що дорівнює порогові коагуляції, відповідає потенціальна крива 4 (рис.10.2). Потенціал поверхні дорівнює деякому критичному (25÷40 мВ), а не ізоелектричному стану.
Оскільки при специфічній адсорбції можлива перезарядка, то для нейтралізаційної коагуляції характерна наявність другої області стійкості: якщо концентрація електроліту перевищить певний необхідний для коагуляції мінімум, частинки змінять знак (перезарядяться) і система знову стане стійкою.
Такий тип коагуляції спостерігається при додаванні неіндиферентних електролітів, що можуть утворювати нерозчинні сполуки з поверхнею частинок, а також індиферентних електролітів, здатних до специфічної адсорбції. (Див. розділ «Вплив електролітів на ПЕШ»).
Концентраційна коагуляція характерна для сильно заряджених золів і суспензій (φ0>100 мВ). В цьому випадку коагуляція зумовлена стисканням λ-шару ПЕШ в результаті збільшення іонної сили розчину. Вона спостерігається при додаванні індиферентних електролітів нездатних до специфічної адсорбції.
За теорію ДЛФО поріг коагуляції сильно заряджених золів не залежить від величини потенціалу частинки і обернено пропорційний шостому степеню заряду іона коагулятора:
|
c |
3 (k T )5 |
(10.5) |
|
A2 |
(e z)6 |
|||
|
|
або
z16 (10.6)
Отже пороги коагуляції одно-, двота тривалентних іонів відносяться як: γ1: γ2 : γ3 = 1 : 1/26 : 1/36 = 1 : 1/64 : 1/729 (10.7)
Дане відношення погоджується з експериментальними даними і дає теоретичне обґрунтування емпіричному правилу Шульце-Гарді: коагулюючу дію чинить протиіон, а коагулююча здатність зростає при збільшенні заряду протиіона.
© Ю.Павловський 2011

58
Недоліки теорії ДЛФО:
1)не враховує природи іонів однакового заряду. Відповідно до рівняння (10.5) поріг
коагуляції однаковий для усіх іонів ліотропного ряду:
Li+ < Na+ <NH4+< K+ < Rb+ < Cs+.
Насправді він зменшується від Li+ до Cs+, що можна пояснити специфічною адсорбцією.
2)не пояснює коагулюючу дію суміші електролітів, оскільки в цьому випадку в досліді спостерігається:
адитивність – дія електролітів підсумовується;
антагонізм – один електроліт послаблює дію іншого;
синергізм – один електроліт підсилює дію іншого.
Коагуляцію використовують для очистки питної води від природних зависей, промислових та побутових стоків від забруднень, для отримання багатьох продуктів харчування, для зупинки кровотечі тощо.
Кінетика коагуляції
Розрізняють швидку коагуляцію, при якій кожне зіткнення частинок приводить до їх злипання і швидкість не залежить від концентрації електроліту і повільну коагуляцію, швидкість якої залежить від концентрації. На графіку залежності w=f(c) можна виділити три області (рис.10.3).
Рис. 10.3. Залежність швидкості коагуляції від концентрації електроліту: 1) область стійкості, 2) область повільної коагуляції; 3) область швидкої коагуляції.
В точці А концентрація відповідає порогу коагуляції , тобто найменшій концентрації, при якій спостерігається коагуляція; ζ-потенціал має критичне значення.
Точка В характеризує «коагулююче число» - таку концентрацію електроліту, при якій швидкість коагуляції максимальна, а ζ-потенціал дорівнює нулю, кожне зіткнення частинок приводить до їх злипання.
Теорію кінетики швидкої коагуляції розробив польський вчений Смолуховський, повільної – російський учений Фукс.
За теорією Смолуховського на швидкість швидкої коагуляції в умовах колоїдної системи впливають три основних фактори:
інтенсивність броунівського руху, мірою якого є коефіцієнт дифузії D;радіус сфери притягання частинок ρ;
початкова концентрація частинок n0.
© Ю.Павловський 2011

59
Механізм коагуляції уявляється наступним чином: частинки золю, що були у вихідній системі до початку коагуляції стикаються одна з одною і утворюють подвійні частинки, ті стикаючись одна з одною, або з початковими, дають частинки кратності 3,4 і т.д. Тобто, механізм подібний до бімолекулярної реакції, а швидкість пропорційна квадрату коагуляції частинок:
w dn kn2 (10.8) d
Константа швидкості коагуляції враховує коефіцієнт дифузії (D) і радіус сфери притягання (ρ) і
дорівнює:
k 4 D (10.9)
З кінетичних даних для другого порядку: |
|
|
|
|
|
|
|
|
|
|
|||
|
|
1 |
|
|
1 |
|
|
|
1 |
|
|
|
|
k |
|
|
|
|
|
|
|
|
|
|
(10.10) |
||
|
|
|
|
|
|||||||||
|
|
|
n |
|
|
n |
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
0 |
|
||
або |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
n0 |
|
1 |
|
|
(10.11) |
|||||||
|
n |
|
|
|
|||||||||
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
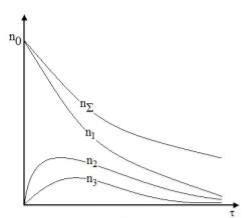
Рівняння (10.11) є лінійним в координатах n0/nΣ=f(τ), а кутовий коефіцієнт даного рівняння tgα=1/ θ, де θ – час половинної коагуляції (Рис.10.3).
Рис.10.4. Графічне знаходження часу половинної коагуляції
Знайшовши час половинної коагуляції θ, можна розрахувати константу коагуляції
kексп. |
1 |
(10.12) |
|
|
|||
n0 |
|||
|
|
і порівняти її з теоретичним значенням, яке можна знайти за наближеною формулою, яка виведена з рівняння (10.9) підстановкою коефіцієнта дифузії D та заміною ρ≈2r:
kтеор. 4 kb T (10.13) 3
Теорія Смолуховського дає рівняння для знаходження кількості частинок певної кратності на будь-який момент часу від початку коагуляції:
ni |
|
i 1 |
|
(10.14) |
||
(1 |
)i |
1 |
||||
|
|
|
||||
© Ю.Павловський 2011
60
Рис. 10.5. Залежність загальної кількості частинок та частинок кратності і=1;2;3 від часу при швидкій коагуляції
Колоїдний захист
Колоїдним захистом називають підвищення агрегативної стійкості дисперсних систем в результаті додавання захисних речовин.
Речовинами здатними до колоїдного захисту у водному середовищі є:білки: желатин, казеїн, альбумін, гемоглобін та ін.;вуглеводи: декстрин,крохмаль, целюлоза, лігнін та ін.;жирні кислоти та їх солі – мила (ПАР).
Для органічного середовища можна використати каучук і знову ж таки, ПАР.
Додавання захисної речовини до золю надає йому властивостей розчину цієї речовини. Наприклад, захищений білком золь оксиду срібла (протаргол) можна випарити насухо і знову перевести в колоїдний стан додаванням води. Концентрування ж золю оксиду срібла без захисної речовини неминуче приводить до його коагуляції.
Захищені золі не підпорядковуються правилу Шульце-Гарді, їх електрофоретична рухливість рівна рухливості захисної речовини.
Механізм захисної дії можна пояснити утворенням навколо дисперсної частинки адсорбційної оболонки з молекул високомолекулярної сполуки. Ця оболонка забезпечує сольватацію частинки і досить великий ζ-потенціал.
Стабілізуючу дію можна також пояснити тим, що шар адсорбованих молекул не дає можливості колоїдними частинкам зблизитись на таку відстань, на якій переважає притягання між ними. Колоїдний захист використовують при виготовленні:
емульсійних фарб, лаків, технічних емульсій;будівельних розчинів (суспензій);каталізаторів;
харчових продуктів: майонез та інші соуси, креми, тісто…;косметичних засобів: пінки, креми, пудри, туші, помади…;лікарських препаратів: емульсій, мазей, суспензій, кремів, золів…
© Ю.Павловський 2011