


2.ЛАБОРАТОРНЫЕ РАБОТЫ
2.1.Простыереакции
Лабораторная работа 1
Изучениескоростигидролизауксусногоангидрида методомэлектрическойпроводимости
Цельизадачиработы
Цель работы – определение скорости гидролиза уксусного ангидрида по изменению электропроводности раствора в ходе реакции. В задачи работы входят ознакомление с методом электрической проводимости, графическое определение константы скорости гидролиза, постоянных в уравнении Аррениуса, расчет теплоты и энтропии активации.
Теоретическоевведение
Гидролиз уксусного ангидрида описывается уравнением (СН3СО)2О + Н2О ↔ 2СН3СООН
и относится к бимолекулярным реакциям. В случае применения неводных растворителей (уксусная кислота, ацетон и т.п.) для оценки скорости реакции пользуются кинетическим уравнением второго порядка:
,
где k – константа скорости реакции; Су.а – концентрация уксусного ангидрида, моль/л; Сw – молярная концентрация воды, моль/л.
Очевидно, что в разбавленных водных растворах молярная концентрация воды много больше молярной концентрации уксусного ангидрида, следовательно, уменьшением Сw в ходе процесса можно пренебречь и значение ее считать постоянным, включив в константу скорости. Таким образом, при указанных условиях скорость процесса будет пропорциональна только концентрации ангидрида и реакция может быть отнесена к классу псевдомолекулярных. Тогда кинетическое уравнение примет вид
dC k Cy.a .
d
Константа скорости может быть рассчитана по уравнению
Химическая кинетика. Лаб. практикум |
23 |

2.ЛАБОРАТОРНЫЕ РАБОТЫ
2.1.Простые реакции
k |
1 |
ln |
|
|
C0 |
, |
|
C |
0 |
C |
|||
|
|
|
|
|
|
где С0 – начальная концентрация уксусного ангидрида, моль/л; ∆С – изменение концентрации уксусного ангидрида, моль/л, за время τ, с.
Кинетику данной реакции наиболее удобно изучать методом электрической проводимости, не требующим отбора проб для анализа. Поскольку образующееся вещество – уксусная кислота – является электролитом, то по мере протекания реакции (т.е. накопления в растворе кислоты) электропроводность системы увеличивается вследствие диссоциации СН3СООН. На начальном этапе из-за низких концентраций продукта непостоянством степени диссоциации уксусной кислоты можно пренебречь. Количество образующейся кислоты пропорционально количеству уксусного ангидрида. Концентрация СН3СООН определяется по электрической проводимости раствора, измеряемой при помощи кондуктометра.
Если обозначить удельную электропроводность раствора в начальный момент реакции χ0, а в какой-то момент времени χτ и в конце реакции χ∞ (когда она уже не меняется), тогда начальная концентрация уксусного ангидрида C0 const ( 0 ) , а концентрация его в какой-то момент времени
C const ( ) и уравнение для расчета константы скорости может быть записано в виде
k1 ln 0 .
Зависимость скорости реакции от температуры описывается уравнением Аррениуса:
k A e( Ea  RT )
RT )
или
ln k ln A RETa ,
где k – константа скорости реакции; А – эмпирическая постоянная, называемая предэкспоненциальным множителем; Еа – энергия активации; R – универсальная газовая постоянная; Т – термодинамическая температура.
Энергию активации можно вычислить, если известны значения констант скорости при двух температурах:
ln k2 |
Ea |
|
|
1 |
|
|
|||||||
|
1 |
|
, |
|
|||||||||
|
|
|
|
||||||||||
откуда |
k1 |
|
R T1 |
|
T2 |
|
|||||||
|
|
|
RT1T2 |
|
ln k2 . |
|
|||||||
E |
|
|
|
|
(1) |
||||||||
|
|
|
|||||||||||
|
a |
|
T |
T |
|
k |
|
||||||
|
|
|
2 |
1 |
|
|
|
1 |
|
||||
Предэкспоненциальный множитель вычисляют, используя уравнение |
|||||||||||||
|
|
|
|
|
|
|
|
|
|
||||
Химическая кинетика. Лаб. практикум |
|
|
|
24 |
|||||||||

2.ЛАБОРАТОРНЫЕ РАБОТЫ
2.1.Простые реакции
ln A ln k |
Ea |
. |
(2) |
|
|||
|
RT |
|
|
Меньшую ошибку в значении Еа получают при построении графической зависимости ln k – 1/T для значений константы скорости, измеренных при разных температурах. Угловой коэффициент зависимости равен –Еа/R, а свободный член – ln А.
По полученным в ходе работы данным рассчитывают теплоту активации ∆H≠:
H |
E |
RT , |
|
|
|
(3) |
|||
|
|
|
a |
|
|
|
|
|
|
а также энтропию активации ∆S≠: |
|
|
|
|
|
|
|
|
|
k T |
h |
e |
S |
R |
e |
E |
RT |
, |
(4) |
k e б |
|
|
|
а |
|
||||
где χ – трансмиссионный коэффициент, учитывающий вероятность того, что система, достигнув переходного состояния, пройдет через него в направлении образования продуктов (для большинства реакций его принимают равным 1); kб – константа Больцмана; h – постоянная Планка.
Экспериментальнаячасть
Реактивы, приборы, посуда: уксусный ангидрид, установка для изучения электропроводности, включающая кондуктометр «Мультитест», колба объемом 50 мл, секундомер.
Последовательность выполнения работы. Предварительно необхо-
димо ознакомиться с правилами эксплуатации кондуктометра «Мультитест»
(см. прил. 2).
Эксперимент, результаты которого используют для получения константы скорости и последующего расчета энергии активации, необходимо проводить в термостатических условиях.
Термостат устанавливают на указанную преподавателем температуру, проверяют постоянство температурного режима (допустимые колебания – 0,1–0,2 оС), подготавливают кондуктометр к работе.
После того как подготовительные процедуры выполнены, в мерную колбу объемом 50 мл помещают 6 мл уксусного ангидрида, объем раствора доводят дистиллированной водой до метки. Вода должна быть предварительно термостатирована. В момент начала растворения ангидрида включают секундомер и не выключают его до конца опыта (до установления постоянного значения электрической проводимости). Отмечают время начала и конца растворения по исчезновению мути при энергичном встряхивании (после приливания воды к уксусному ангидриду появляется граница раздела двух жидких
Химическая кинетика. Лаб. практикум |
25 |

2.ЛАБОРАТОРНЫЕ РАБОТЫ
2.1.Простые реакции
фаз, при взбалтывании наблюдается помутнение), среднее время принимается за время начала реакции.
Сосуд для измерения электрической проводимости после двукратного ополаскивания исследуемым раствором заполняют так, чтобы электроды были погружены на 0,5–1,0 см ниже уровня раствора. Сосуд помещают в термостат, в котором встряхивают его в течение 3 мин до установления постоянной температуры.
В работе участвуют два человека. Один измеряет электрическую проводимость, второй следит за временем и записывает показания секундомера и кондуктометра. Чем выше температура опыта, тем чаще проводятся измерения. Первые три измерения проводят через 30 с, затем четыре–пять измерений через 1 мин, два–три измерения через 5 мин, далее делаются измерения через 10 мин, через 1 ч и до установления постоянного значения электропроводности.
Результаты измерений заносят в табл. 1.
Таблица 1
Температура опыта, оС … Реакционный объем, мл …
Концентрация уксусного ангидрида, моль/л … Удельная электропроводность в начале опыта, См/см … Удельная электропроводность в конце опыта, См/см …
№ измерения |
τ, с |
χτ |
χ∞ – χτ |
ln(χ∞ – χτ) |
k |
1 |
τ1 |
χ1 |
… |
… |
… |
2 |
τ2 |
χ2 |
… |
… |
… |
n |
τn |
χn |
… |
… |
… |
|
|
|
|
|
kср = |
Для определения константы скорости строят график в координатах ln(χ∞ – χτ) – τ. По отрезку, отсекаемому прямой (после ее экстраполяции) на оси ординат, находят ln(χ∞ – χ0). Вычислив k для каждого момента, определяют среднее значение kср и сравнивают со средним значением, полученным графически (тангенс угла наклона прямой к оси τ).
По константам скорости при двух температурах (второе значение константы берут из справочника) рассчитывают энергию активации данного процесса по уравнению (1) и значение предэкспоненциального множителя по уравнению (2).
По полученным данным вычисляют теплоту (уравнение (3)) и энтропию активации (уравнение (4)) исследованной реакции.
Для подготовки к защите лабораторной работы можно воспользоваться следующей литературой: [1, c. 4–19, 25–32; 4, c. 15–17, 24–27, 46–50; 12, c. 32–36, 43–46; 16, c. 9–19].
Химическая кинетика. Лаб. практикум |
26 |

2.ЛАБОРАТОРНЫЕ РАБОТЫ
2.1.Простые реакции
Контрольныевопросы
1.Что называют скоростью химической реакции?
2.В чем заключается физический смысл константы скорости реакции?
3.Что представляет собой кинетическое уравнение? Почему говорят о «формально-кинетическом уравнении»?
4.Какова размерность константы скорости химической реакции?
5.Что понимают под порядком химической реакции?
6.Есть ли разница между порядком и молекулярностью реакции? Если есть, то в чем она заключается?
7.Почему реакцию гидролиза уксусного ангидрида относят к псевдомолекулярным?
8.Что называют теплотой активации? Как рассчитать энтропию акти-
вации?
9.Правомерно ли использование такого свойства растворов, как электропроводность, для изучения кинетических закономерностей реакции гидролиза уксусного ангидрида? Почему?
10.Чем обусловлена электрическая проводимость растворов? Какие факторы влияют на величину электропроводности раствора?
Лабораторная работа 2
Изучениекинетикиреакциигидролиза уксусногоангидридаколориметрическимметодом
Цельизадачиработы
Цель работы – определение скорости гидролиза уксусного ангидрида колориметрическим методом. В задачи работы входят ознакомление с колориметрическим методом и эксплуатацией спектрофотометра «SPEKOL-1300», графическое определение константы скорости гидролиза, расчет энергии активации при заданной температуре.
Теоретическоевведение
В основе метода лежит окислительно-восстановительная реакция, протекающая между йодид- и йодат-ионами:
8I- + IO3- + 6H+ → 3I3- + 3H2O
Поскольку указанная реакция может протекать только в кислой среде, то количество выделившегося йода будет связано с количеством образовавшейся уксусной кислоты при гидролизе уксусного ангидрида:
Химическая кинетика. Лаб. практикум |
27 |

2.ЛАБОРАТОРНЫЕ РАБОТЫ
2.1.Простые реакции
(СН3СО)2О + Н2О ↔ 2СН3СООН
Необходимо отметить, что в данном случае константой диссоциации кислоты пренебрегать нельзя, поскольку расчет ведется с использованием измерений по сопряженной реакции, скорость которой, очевидно, определяется концентрацией Н+. Значения Кд при некоторых температурах приведены в табл. 2.
|
|
|
Таблица 2 |
|
|
|
|
|
|
t, оC |
Kд·105 |
t, оC |
Kд·105 |
|
0 |
1,66 |
30 |
1,75 |
|
10 |
1,73 |
35 |
1,73 |
|
20 |
1,75 |
40 |
1,70 |
|
25 |
1,75 |
|
|
|
Реакция гидролиза уксусного ангидрида в водном растворе является, как уже указывалось в предыдущей работе, псевдомолекулярной, поэтому скорость ее может быть выражена уравнением
dCy.к k Cу.а0 Cу.к ,
d
где Су.к – общая концентрация образовавшейся в результате гидролиза уксусной кислоты; Су.а 0 – исходная концентрация уксусного ангидрида; k – константа скорости реакции гидролиза; τ – время реакции. Интегрирование уравнения дает выражение
k |
1 |
ln |
Cу.а0 |
. |
|
|
Су.а0 Су.к |
||||
|
|
|
Скорость йодид-йодатного взаимодействия можно описать уравнением
ddCI k1 CH2 CI0 CI 3 ,
где СI – количество образовавшегося в результате реакции йода; СН+ – концентрация ионов Н+, образовавшихся при диссоциации уксусной кислоты; СI 0 – концентрация йода в исходной йодид-йодатной смеси, пересчитанная на атомарный I; k1 – константа скорости реакции окисления I-; τ – время реакции. После интегрирования получается
Химическая кинетика. Лаб. практикум |
28 |

2.ЛАБОРАТОРНЫЕ РАБОТЫ
2.1.Простые реакции
2 |
|
1 |
|
1 |
|
|
1 |
|
|
С |
|
|
|
|
|
|
|
|
. |
2k1 |
|
CI |
|
CI0 |
|||||
H |
|
CI0 |
2 |
|
|
||||
|
|
|
|
|
|
2 |
|
||
Поскольку величина Су.к включает содержание ацетат-ионов и недиссоциированной уксусной кислоты, а количество свободного йода соответствует количеству СН3СОО--ионов, можно записать
Су.к CH3COO СН3СООН СI СН3СООН ,
а для константы диссоциации
Kд |
[H ] [CH COO ] |
|
C |
CI |
|
||
3 |
|
H |
|
|
. |
||
[CH3COOH] |
[CH3COOH] |
||||||
|
|
|
|||||
Отсюда
CH3COOH CH CI ,
Kд
Cу.к СI CH CI .
Kд
Таким образом, количество образующегося йода (в данном случае пересчитанное на атомарный йод) может быть использовано в качестве параметра для определения скорости реакции, поскольку установить его в реакционной смеси колориметрически можно в любой момент времени.
Значения константы скорости k1 реакции окисления I- йодат-ионом для некоторых температур приведены в табл. 3.
Таблица 3
|
k1·10-10 |
t, оC |
|
0 |
2,54 |
15 |
4,80 |
25 |
6,00 |
Химическая кинетика. Лаб. практикум |
29 |

2.ЛАБОРАТОРНЫЕ РАБОТЫ
2.1.Простые реакции
Экспериментальнаячасть
Реактивы, приборы, посуда: раствор йода (I2) 0,1 М, уксусный ангидрид, раствор KIO3, раствор KI, колбы объемом 50 и 100 мл, спектрофотометр
«SPEKOL-1300», секундомер.
Последовательность выполнения работы. Подготавливают спектро-
фотометр к измерениям (инструкция по эксплуатации прибора приведена в прил. 3).
Готовят серию стандартных растворов йода (5·10-4, 1·10-3, 2·10-3, 7,5·10-3, 1,2·10-2, 2,5·10-2 моль/л) разбавлением 0,1 М раствора I2.
В работе используют кювету толщиной 10 мм.
Проводят измерения оптической плотности стандартных растворов, в качестве раствора сравнения используют дистиллированную воду (см. прил. 3, п. 4). Результаты измерений оформляют в виде табл. 4.
|
|
|
Таблица 4 |
||
|
|
|
|
|
|
Длина волны, нм … |
|
|
|
||
Толщина кюветы, мм … |
|
|
|
||
Температура, оС … |
|
|
|
||
№ колбы |
Объем 0,1 М раствора I2 |
Концентрация I2, |
Оптическая плот- |
|
|
для разбавления, мл |
моль/л |
ность А |
|||
|
|||||
|
|
|
|
|
|
Для удобства дальнейших расчетов концентрацию йода пересчитывают на атомарный йод (путем удвоения полученной концентрации I2). По полученным данным строят калибровочный график в координатах «Оптическая плотность – Концентрация I».
После получения графика готовят 100 мл раствора, содержащего 0,02 моль/л KIO3 и 0,16 моль/л KI. На аналитических весах взвешивают 0,06 мл уксусного ангидрида (объем можно варьировать от 0,2 до 0,01 мл) и переносят навеску в мерную колбу (V = 50 мл), в которую предварительно наливают 30 мл приготовленного раствора смеси KI и KIO3. Доводят объем до метки тем же йодид-йодатным раствором.
В момент сливания уксусного ангидрида с раствором KI и KIO3 включают секундомер и не выключают его до окончания эксперимента. Замечают время добавления уксусного ангидрида и время конца растворения (момент гомогенизациираствора), среднеевремяпринимаютзавремяначалареакции.
Полученную реакционную смесь помещают в кювету. В качестве раствора сравнения используют раствор йодида и йодата калия. По мере протекания реакции проводят измерения оптической плотности раствора А: первые три измерения через каждые 30 с, затем пять измерений через 1 мин, еще 3 измерения через 5 мин, затем через 10 и 20 мин до установления постоянного значения А, что свидетельствует об окончании реакции.
Химическая кинетика. Лаб. практикум |
30 |
2.ЛАБОРАТОРНЫЕ РАБОТЫ
2.1.Простые реакции
По оптической плотности раствора при помощи калибровочного графика определяют концентрацию атомарного йода. Исходную концентрацию уксусного ангидрида в растворе рассчитывают с учетом навески.
Полученные данные оформляют в виде табл. 5.
Таблица 5
Температура, оС …
m (СН3СО)2О), г … Су.а, моль/л … СI 0, моль-экв/л … k1 …
Время |
А |
СI, |
СI 0 – СI, |
СН+ · 106, |
Су.к, |
[у.а.], |
k |
||
измере- |
от начала |
||||||||
моль/л |
моль-экв/л |
моль/л |
моль/л |
моль/л |
|||||
ния |
реакции, с |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Для определения константы скорости строят график в координатах ln[у.а.] – τ ([у.а.] – равновесная концентрация уксусного ангидрида). Вычислив k для каждого момента, определяют среднее значение kср и сравнивают со средним значением, полученным графически (тангенс угла наклона прямой к оси τ).
Энергию активации рассчитывают по константам скорости при двух температурах (вторую константу берут из справочника) по уравнению
E |
|
|
RT1T2 |
|
ln k2 . |
|
|
|
|
||||
|
a |
|
T |
T |
k |
|
|
|
|
2 |
1 |
|
1 |
Для подготовки к защите лабораторной работы можно воспользоваться следующей литературой: [1, c. 4–19, 25–32; 4, c. 15–17, 24–27, 46–50; 12, c. 32–36, 43–46; 16, c. 9–19].
Контрольныевопросы
1.На чем основано изучение кинетики реакции гидролиза уксусного ангидрида колориметрическим методом?
2.В чем заключается сущность графического метода определения константы скорости реакции?
3.Что называют энергией активации? Отличается ли ее величина от величины энергии активированного комплекса? Почему?
4.Почему при использовании колориметрического метода нельзя пренебрегать константой диссоциации уксусной кислоты?
5.Какие факторы влияют на величину оптической плотности изучаемых растворов?
6.Каков характер зависимости оптической плотности раствора от концентрации йода? Почему?
7.Отличаются ли значения кинетических характеристик, полученные разными методами? Если отличаются, то почему?
Химическая кинетика. Лаб. практикум |
31 |

2.ЛАБОРАТОРНЫЕ РАБОТЫ
2.2.Сложныереакции
Лабораторная работа 3
Изучениекинетикиреакцииокислениятиомочевины
итиоацетамидаионамигексацианоферрата(III)
вщелочномрастворе
Цельизадачиработы
Цель работы – определение константы скорости окисления тиомочевины и тиоацетамида ионами гексацианоферрата (III) в щелочном растворе. В задачи работы входят ознакомление с эксплуатацией спектрофотометра «SPEKOL-1300», определение константы скорости окисления тиомочевины и тиоацетамида, расчет энергии активации при заданной температуре.
Теоретическоевведение
Реакции окисления тиомочевины и тиоацетамида ионами гексацианоферрата (III) могут быть описаны следующими уравнениями:
(NH2)2CS + 10OH- + 8[Fe(CN)6]3- ↔ (NH2)2CO + SO42- + 8[Fe(CN)6]4- + 5H2O
CH3CSNH2 + 11OH- + 8[Fe(CN)6]3- ↔ CH3COO- + SO42- + NH3 + + 8[Fe(CN)6]4- + 5H2O
Скорости обеих реакций зависят от концентрации гидроксид-ионов, что свидетельствует об образовании промежуточной енольной формы. В связи с чем предлагается следующий механизм рассматриваемых реакций:
1 стадия – образование енольного промежуточного аниона – обратима, скорость зависит от природы восстановителя:
|
k |
|
|
|
|
RCSNH2 ОН |
1 |
RCSNH Н2О |
|
||
|
k2 |
|
2 стадия – образование промежуточного комплекса с окислителем:
RCSNH-+ [Fe(CN)6 ]3 k2 Комплекс
3 стадия – превращение промежуточного комплекса в конечный продукт реакции или следующий промежуточный продукт, который сразу переходит в конечный продукт:
Химическая кинетика. Лаб. практикум |
32 |

2.ЛАБОРАТОРНЫЕ РАБОТЫ
2.2.Сложные реакции
Комплекс + [Fe(CN)6 ]3 k3 2[Fe(CN)6 ]4 Продукт.
Скорость последней стадии значительно больше предыдущих.
Сделав предположение, что концентрация промежуточного енольного аниона RCSNH- стационарна, в соответствии с методом стационарных концентраций получают
d[RCSNH ] k1 [RCSNH2 ] [OH ] k1 [RCSNH ] d
k2 [RCSNH ] [Fe(CN)36 ] 0,
[RCSNH ] k1 [RCSNH2 ] [OH ] . k1 k2 [Fe(CN)36 ]
Скорость реакции также может быть выражена через концентрацию гексацианоферрата (III):
d[Fe(CN)36 ] k |
2 |
[RCSNH ] [Fe(CN)3 ] k |
3 |
[Комплекс] [Fe(CN)3 ]. |
d |
6 |
6 |
||
|
|
|
|
С учетом того, что, согласно предложенному механизму, в реакции окисления одного моля RCSNH2 участвуют два моля [Fe(CN)6]3-, получается
|
d[Fe(CN)3 ] |
|
2k |
k |
2 |
[RCSNH |
|
] [OH ] [Fe(CN)3 ] |
|
||
|
6 |
|
1 |
|
|
|
|
2 |
6 |
. |
|
d |
|
|
|
k |
|
||||||
|
|
|
|
|
k |
2 |
[Fe(CN)3 ] |
|
|||
|
|
|
|
|
|
1 |
|
|
6 |
|
|
В реакции окисления тиомочевины лимитирующей является стадия 2, |
||||||
поэтому k1 >> k2 и уравнение для скорости можно представить в виде |
||||||
|
d[Fe(CN)36 |
|
2k1k2 |
|
|
3 |
|
d |
|
|
[NH2CSNH2 ] [OH |
|
] [Fe(CN)6 ]. |
|
|
k1 |
|
|
|
|
Таким образом, окисление тиомочевины щелочным раствором, содержащим ионы гексацианоферрата (III), условно характеризуется первым порядком по каждому из участников реакции.
В реакции окисления тиоацетамида скорость определяется первой ста- |
|||||
дией, поэтому k1 |
>> k2 и k1 << k1 : |
|
|
|
|
|
d[Fe(CN)36 |
2k |
[СH CSNH |
2 |
] [OH ]. |
|
d |
1 |
3 |
|
|
|
|
|
|
|
|
Видно, что скорость реакции окисления тиоацетамида не зависит от концентрации гексацианоферрат(III)-ионов и имеет нулевой порядок по данному компоненту. Условный порядок реакции по тиоацетамиду и гидроксидиону равен 1. Необходимо заметить, что при низких концентрациях окисли-
Химическая кинетика. Лаб. практикум |
33 |

2.ЛАБОРАТОРНЫЕ РАБОТЫ
2.2.Сложные реакции
теля лимитирующей становится стадия 2 и расчеты выполняют, как и для случая с тиомочевиной.
Поскольку стадия 2 представляет собой взаимодействие двух отрицательно заряженных ионов, скорость ее зависит от ионной силы раствора и диэлектрической постоянной среды.
Так как на скорости окисления обоих соединений сильно влияет концентрация щелочи, кинетические закономерности этих реакций изучают при постоянном значении рН, которое поддерживают при помощи карбонатного буферного раствора (смесь равных объемов 0,1 М раствора Na2CO3 и 0,1 М раствора NaHCO3), обеспечивающего рН 11.
Положительное значение при изучении кинетики указанных реакций имеет тот факт, что процессы идут с заметной скоростью при нагревании выше 30 оС и замедляются при снижении температуры. Протекание реакции легко контролируется по изменению концентрации ионов [Fe(CN)6]3- в растворе при помощи спектрофотометра «SPECOL-1300»: ион [Fe(CN)6]4-, в отличие от [Fe(CN)6]3-, не поглощает в области 400–450 нм.
Экспериментальнаячасть
Реактивы, приборы, посуда: раствор гексацианоферрата (III) калия (К3[Fe(CN)6]) 0,02 М, карбонатный буферный раствор, раствор тиомочевины 0,02 М, колбы объемом 50 мл (7 шт.), 100 и 200 мл, термостат, спектрофото-
метр «SPEKOL-1300», секундомер.
Последовательность выполнения работы. Подготавливают спектро-
фотометр к измерениям, как указано в прил. 3.
В мерных колбах (объемом 50 мл) готовят серию стандартных раство-
ров К3[Fe(CN)6] (1·10-4, 2·10-4, 2,5·10-4, 3·10-4, 4·10-4, 5·10-4, 6·10-4 моль/л) раз-
бавлением 0,02 М раствора К3[Fe(CN)6] карбонатным буферным раствором (смесь равных объемов 0,1 М раствора Na2CO3 и 0,1 М раствора NaHCO3).
Проводят измерения оптической плотности стандартов (см. прил. 3, п. 4), используя в качестве раствора сравнения карбонатный буфер. Результаты измерений оформляют в виде табл. 6.
|
|
|
Таблица 6 |
|
|
|
|
Длина волны, нм … |
|
|
|
Размер кюветы, мм … |
|
|
|
№ колбы |
Концентрация К3[Fe(CN)6], |
Объем 0,02 М р-ра К3[Fe(CN)6] |
Оптическая |
|
моль/л |
для приготовления стандарта, мл |
плотность |
|
|
|
|
После построения калибровочного графика в термостат с заданной температурой (не ниже 35 оС) помещают мерную колбу (объемом 500 мл) с 10–40 мл 0,02 М раствора К3[Fe(CN)6] в буферной смеси; колбу со 100–400 мл
Химическая кинетика. Лаб. практикум |
34 |

2.ЛАБОРАТОРНЫЕ РАБОТЫ
2.2.Сложные реакции
0,02 М раствора тиомочевины (или тиоацетамида, в зависимости от задания) в буферной смеси; колбу с карбонатным буферным раствором.
Объемы растворов реагентов рассчитываются исходя из того, что концентрация тиомочевины в реакционной смеси должна быть в 10 раз больше концентрации гексацианоферрата (III) калия (в случае тиоацетамида концентрации реагентов могут быть близкими по значению). Высокая концентрация окислителя может привести к образованию элементной серы и, как следствие, к помутнению раствора. Нижний предел концентрации К3[Fe(CN)6] определяется чувствительностью спектрофотометра. При содержании [Fe(CN)6]3- выше 6·10-3 моль/л раствор перед определением разбавляют.
После выравнивания температур раствор тиомочевины (тиоацетамида) приливают в колбу с раствором гексацианоферрата (III) калия. Отмечают время сливания и считают его временем начала реакции. Объем раствора доводят до метки термостатированным буфером. Колбу с реакционной смесью помещают в термостат.
По ходу реакции из реакционной смеси отбирают пробы, которые быстро охлаждают водой со льдом и колориметрируют. Концентрацию гексацианоферрата определяют по значениям оптической плотности при помощи калибровочного графика (см. прил. 3, п. 5). Первые две пробы отбирают с интервалом 5 и 10 мин от начала реакции, затем – в зависимости от характера изменения оптической плотности раствора. Объем проб зависит от размера выбранной кюветы.
Полученные результаты заносят в табл. 7.
|
|
|
|
|
Таблица 7 |
|
|
|
|
|
|
|
|
Температура, оС … |
|
|
|
|
||
Исходная концентрация К3[Fe(CN)6], моль/л … |
|
|
|
|||
Исходная концентрация тиомочевины (тиоацетамида), моль/л … |
|
|
||||
|
Время |
А |
Концентрация |
k |
||
измерения |
|
от начал реакции, с |
К3[Fe(CN)6], моль/л |
|||
|
|
|
|
|||
|
|
|
|
|
|
|
Поскольку концентрация гидроксид-ионов в ходе реакции практически не изменяется, а количество тиомочевины на порядок больше количества К3[Fe(CN)6], содержание тиомочевины можно считать постоянным и константу скорости вычислить по уравнению для реакций первого порядка:
d[Fe(CN)36 ] |
k [Fe(CN)36 ]. |
d |
|
По значениям констант скоростей при двух температурах (вторую константу берут из справочника) рассчитывают энергию активации реакции.
Для подготовки к защите лабораторной работы можно воспользоваться следующей литературой: [1, c. 4–19, 25–54; 4, c. 15–43, 46–50; 12, c. 32–36, 43–61; 16, c. 9–19, 47–91].
Химическая кинетика. Лаб. практикум |
35 |

2.ЛАБОРАТОРНЫЕ РАБОТЫ
2.2.Сложные реакции
Контрольныевопросы
1.Какие реакции называют сложными? Как классифицируются сложные реакции? К какому типу относятся изучаемые в работе реакции?
2.Чем обусловлена разница кинетических уравнений для реакций окисления тиомочевины и тиоацетамида?
3.В чем заключается принципиальное отличие простых и сложных ре-
акций?
4.Чем вызвана необходимость использования буферного раствора при изучении кинетики рассматриваемых в работе реакций?
5.На чем основан метод колориметрического изучения кинетики окисления тиомочевины (тиоацетамида) К3[Fe(CN)6]?
6.Какое допущение используется в методе стационарных концентраций? В каких случаях данный метод может быть применен?
7.На каком основании при расчете кинетических характеристик реакции окисления тиомочевины используют уравнение для реакции первого порядка? Правомерно ли это?
2.3. Влияниетемпературы наскоростьреакции
Лабораторная работа 4
Изучениекинетикиокисленияметалловисплавов
Цельизадачиработы
Цели работы – установление закона окисления металла или сплава и определение кинетических параметров этого процесса. В задачи работы входит определение константы скорости и энергии активации окисления в условиях газовой коррозии.
Теоретическоевведение
Примером газовой коррозии металлов может служить окисление последних при высоких температурах. При комнатной температуре окисление металлов чаще всего ограничивается потускнением или образованием тонких пленок. Вследствие малой диффузии кислорода к поверхности металла при таких температурах толщина этих пленок практически не увеличивается. Особое положение в условиях химических производств имеет газовая корро-
Химическая кинетика. Лаб. практикум |
36 |

2.ЛАБОРАТОРНЫЕ РАБОТЫ
2.3.Влияние температуры на скорость реакции
зия при высоких температурах. Такая коррозия является равномерной, т.к. распространяется вглубь металла почти одинаково по всей поверхности, а образующиеся пленки из продуктов коррозии имеют одинаковую толщину по всей площади, подвергающейся коррозии.
Скорость газовой коррозии оценивается по привесу с квадратного метра поверхности в единицу времени или по глубине коррозии.
При повышении температуры скорость коррозии возрастает, т.к. увеличивается скорость диффузии и химических процессов. Вследствие этого пленки продуктов коррозии утолщаются. Увеличение толщины пленки на разных металлах протекает по различным законам: прямолинейному, параболическому и логарифмическому. Данное обстоятельство обусловливается преимущественно природой образующихся оксидов, а также целостностью покрытия. У металлов, на которых при окислении не образуется защитная пленка, скорость роста остается постоянной. Ее толщину рассчитывают на основании формулы
dd k C ,
т.е.
k A,
где δ – толщина слоя продукта коррозии; kδ – коэффициент скорости процесса; C – движущая сила коррозии; А – постоянная интегрирования.
Константа интегрирования определяет толщину пленки в начальный момент окисления, т.е. τ = 0. Если окисление начинается на чистой поверхности, то А = 0.
Ряд металлов, применяемых в химической технологии, окисляется по параболическому закону, например, металлы, на которых в результате химической коррозии получаются сплошные пленки. В этом случае процесс тормозится диффузией агентов через пленку, и по мере ее роста коррозия замедляется.
Представим сплошную пленку толщиной δ, находящуюся на поверхности окисляющегося металла (рис. 2).
С0 С1 Ме δ
О2
Рис. 2. Схема диффузии кислорода через пленку при соприкосновении металла
Для этого случая имеем соотношение
Химическая кинетика. Лаб. практикум |
37 |

2.ЛАБОРАТОРНЫЕ РАБОТЫ
2.3.Влияние температуры на скорость реакции
dG |
DF dC , |
|
d |
|
d |
где G – количество диффундирующего вещества; τ – время; D – коэффициент |
||
диффузии; F – площадь окисления; |
dC |
– градиент концентрации. |
|
||
|
d |
|
Если принять, что в процессе окисления устанавливается стационарный режим диффузии, т.е. не происходит накопления диффундирующего вещест-
ва в каком-либо сечении пленки, то производная |
dC |
заменяется соотноше- |
|||
d |
|||||
|
C0 C1 |
|
|
||
нием |
. Здесь С0 – концентрация кислорода на внешней поверхности |
||||
|
|||||
|
|
|
|
||
пленки на границе с воздухом, С1 – концентрация кислорода на внутренней поверхности пленки на границе с металлом. Тогда скорость диффузии для единичной поверхности пропорциональна разности концентраций кислорода и обратно пропорциональна толщине слоя:
UD dG |
D C0 C1 . |
d |
|
Примем, что на корродируемом металле уже имеется защитная пленка, а процесс идет в диффузионной области, и весь кислород, диффундируя через пленку, не накапливается, т.е. немедленно вступает в реакцию. Для такого случая скорость роста пленки Uкор может быть равна скорости диффузии кислорода UD:
Uкор UD D C0 .
При постоянных условиях окисления (парциальное давление кислорода и температура процесса неизменны, например, при воздействии воздуха, нагретого до определенной температуры)
Uкор d |
kD |
d |
|
или после интегрирования
2 kD A.
В некоторых случаях, например, при наличии в растущей пленке трещин (из-за нагревания или охлаждения), а также при сравнительно невысоких температурах коррозия металлов протекает по логарифмическому закону:
d |
|
k |
d |
|
e |
или
ln k .
Химическая кинетика. Лаб. практикум |
38 |

2.ЛАБОРАТОРНЫЕ РАБОТЫ
2.3.Влияние температуры на скорость реакции
При увеличении температуры скорость окисления возрастает. Это происходит потому, что с повышением температуры увеличивается скорость диффузии кислорода и, как следствие, константа скорости окисления в соответствии с уравнением Аррениуса:
ln k A |
E |
. |
|
|
|
|
|
|
|
||||
|
RT |
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
М+n |
|
|
|
|
|
М |
X-m |
X |
|
|
|
|
|
e- |
|
Рис. 3. Схема движения частиц |
|
|
|
|
|
|
|
|
|
|
X |
|
|
при окислении металла |
|
|
|
|
|
|
Перенос вещества через слой продукта реакции окисления – окалину осуществляется путем независимого движения заряженных частиц: ионов (катионов M+n, X-m) и электронов е-. Согласно схеме, изображенной на рис. 3, катионы и электроны движутся в одном направлении – от границы окалины с металлом к границе с неметаллом, анионы – в противоположном.
Процесс переноса через слой окалины является лимитирующей стадией окисления: переход ионов или электронов через поверхность раздела фаз протекает без затруднений, поэтому на межфазных границах существует термодинамическое равновесие.
Экспериментальнаячасть
Реактивы, приборы, посуда: металлы, заданные преподавателем, пинцет, установка для изучения окисления металлов.
Последовательность выполнения работы. Исследование кинетики газовой коррозии проводится по методике периодического взвешивания на лабораторной установке, схема которой приведена на рис. 4.
6
5 4
3
2
|
|
|
|
7 |
Рис. 4. Схема установки для изучения |
|
|
|
|
|
|||
1 |
|
|
|
скорости газовой коррозии при высоких |
||
|
|
|
|
|
||
|
|
|
|
|
температурах |
|
|
|
|
|
|||
|
|
|
|
|
|
|
Химическая кинетика. Лаб. практикум |
39 |

2.ЛАБОРАТОРНЫЕ РАБОТЫ
2.3.Влияние температуры на скорость реакции
Испытуемый образец 2 подвешивают на нихромовом или кварцевом подвесе 5, состоящем из нескольких звеньев, в вертикальной трубчатой печи сопротивления 3 к коромыслу аналитических весов 6. Температуру в печи замеряют хромель-алюмелевой термопарой 1 и записывают на потенциометре КСП-4 7. Конец термопары должен находиться вблизи испытуемого образца. Чтобы избежать нагрева весов, между печью и весами устанавливается экран 4.
Работа по исследованию скорости окисления металла проводится следующим образом: включают обогрев печи и потенциометр КСП-4.
По достижению заданной температуры (регулируют при помощи ЛАТРа) подвешивают в печи испытуемый образец 2 и через 1–2 мин его взвешивают. За это время образец принимает температуру печи, но еще не успевает заметно окислиться. Далее в течение 1–2 ч через заданные преподавателем промежутки времени продолжают взвешивание образца. По окончании опыта при данной температуре образец вынимают, устанавливают в печи следующую заданную температуру, подвешивают в печи новый образец такого же металла и повторяют опыт в том же порядке.
Результаты опытов по исследованию кинетики газовой коррозии заносят в табл. 8.
По табличным данным для каждой температуры строят графики в координатах G1 – τ, G12 – τ и G1 – ln τ, устанавливая таким образом, по какому закону идет окисление в определенной температурной области.
Таблица 8
Металл … Номер образца …
Поверхность образца, м2 …
№ |
Время |
Масса об- |
|
Привес, кг |
|
взвешивания |
от начала опыта τ |
разца G1, кг |
G1 |
на единицу поверхности G1 |
|
|
|
|
|
|
|
Установив характер кривой и какому уравнению она подчиняется, вычисляют константу скорости окисления при любой температуре, а затем определяют энергию активации и получают расчетное уравнение процесса окисления в зависимости от температуры. Для нахождения константы скорости окисления вычисления проводят по формулам:
–при прямолинейном характере кривых окисления k G1 ;
–при параболическом характере кривых k G12 ;
–при логарифмическом законе окисления k G1 lg .
Для подготовки к защите лабораторной работы можно воспользоваться следующейлитературой: [11, с. 103, 104, 114–118; 16, c. 210–218; 18, с. 387–396].
Химическая кинетика. Лаб. практикум |
40 |
2.ЛАБОРАТОРНЫЕ РАБОТЫ
2.3.Влияние температуры на скорость реакции
Контрольныевопросыизадания
1.Какой можно ожидать закон окисления, если соотношение молярных объемов оксида и металла Vок/Vм < 1?
2.Каким законом описывается процесс окисления, если он лимитируется диффузией в газовой фазе?
3.Напишите уравнения 1-го и 2-го законов диффузии Фика.
4.Расскажите о механизмах окисления различных металлов.
5.Сравните несколько кинетических моделей (3–4) твердофазных реакций с различными лимитирующими стадиями.
6.Перечислите стадии гетерогенного процесса.
7.Запишите линейную форму уравнения Аррениуса и покажите, как графически определить постоянные в этом уравнении.
2.4. Цепныеифотохимические реакции
Лабораторная работа 5
Изучениекинетикифотохимическогоразложения перекисиводорода
Цельизадачиработы
Целью работы является исследование кинетики реакции фотохимического разложения перекиси водорода в водном растворе. В задачи работы входят определение константы нарастания, характеризующей скорость разветвления цепи, и нахождение периода индукции в реакции фотохимического разложения перекиси водорода.
Теоретическоевведение
Реакции, протекающие под действием светового излучения (видимого и ультрафиолетового), которое вызывает активацию частиц одного из реагирующих веществ, называются фотохимическими.
Закон фотохимической эквивалентности был установлен А. Эйнштейном, согласно ему каждая молекула, реагирующая под влиянием света, поглощает только один квант излучения, вызывающий реакцию. Поэтому сис-
Химическая кинетика. Лаб. практикум |
41 |

2.ЛАБОРАТОРНЫЕ РАБОТЫ
2.4.Цепные и фотохимические реакции
тема, в которой прореагировало N молекул, должна поглотить энергию Е в соответствии с уравнением
E Nh Nh C ,
где h – постоянная Планка; ν – частота излучения; С – скорость света; λ – длина его волны.
Обычно число прореагировавших молекул не равняется числу поглощенных квантов. Отношение числа фактически прореагировавших молекул к числу поглощенных квантов γ называется квантовым выходом. Эта величина бывает меньше, больше и равна единице.
Важной особенностью фотохимических реакций является независимость их скорости от температуры. Первичная фотохимическая реакция обычно является лимитирующей, а энергия кванта, поглощенного в ней, много выше энергии теплового движения и изменения ее с температурой.
Цепные реакции были открыты при изучении фотохимических процессов.
Цепные реакции отличаются от обычных тем, что при их протекании элементарные акты не независят друг от друга, а каждый предшествующий акт вызывает один или несколько других актов, т.е. превращение исходных веществ в продукты реакции осуществляется путем регулярного чередования нескольких элементарных реакций. Это связано с тем, что частицы, которые возникают в результате реакции, обладают повышенной химической активностью.
Цепные реакции являются основой многих процессов, имеющих большое практическое значение: горение, крекинг нефти, производство пластических масс (полимеров), атомная энергетика.
Влюбойцепнойреакциипроцессразвиваетсявследствие образования неустойчивых промежуточных продуктов – атомов или радикалов, которые также называются активными центрами. Эти промежуточные вещества являются валентно ненасыщенными. Вследствие неустойчивости атомов и радикалов время ихжизни очень мало.
Возникающие при появлении атомов или радикалов цепи либо неограниченно развиваются, что приводит к взрыву, либо обрываются вследствие гибели активных центров в результате адсорбции на стенках сосуда или тройных соударений в объеме смеси.
Такимобразом, дляцепныхреакцийхарактернытриследующиестадии:
1)зарождение цепи (первичная реакция);
2)развитие цепи;
3)обрыв цепи.
Зарождение цепи не обязательно происходит под влиянием света. Цепные реакции могут возникать под воздействием излучений радиоактивных веществ. В некоторых случаях начало цепных реакций обусловлено добавлением к реакционной смеси веществ, атомы которых поглощают свет определенной длины волны и становятся активными. Возникновение цепей может происходить
Химическая кинетика. Лаб. практикум |
42 |

2.ЛАБОРАТОРНЫЕ РАБОТЫ
2.4.Цепные и фотохимические реакции
ивследствие самого акта химической реакции, при которой образуются радикалы. Кроме того, образование атомов или радикалов может быть связано с высокой температурой реакционной смеси или наличием катализаторов.
Во второй стадии цепных реакций – развитие цепей – можно различать реакции с простыми и с разветвляющимися цепями. В первом случае каждый исчезающий атом или радикал вызывает появление лишь одного нового атома или радикала. Примером такой неразветвленной цепи может служить ре-
акция между Н2 и С12:
Cl2 + hν = 2Cl·
Сl· + Н2 = НС1 + Н· Н· + Сl2 = НС1 + Сl и т.д.
В реакциях с разветвляющимися цепями на каждый исчезающий центр реакции возникает несколько новых. К числу таких реакций, в частности, принадлежит процесс расщепления водорода. При низких давлениях и при температуре около 500 оС он может развиваться согласно следующей схеме:
Н + О2 = ОН + О
OH + Н2 = Н2О+ Н
О + Н2 = ОН + Н и т.д.
Существование свободных атомов и радикалов было доказано различными методами. Для этого в реакционную смесь вводили специальные зонды, снабженные чувствительными малоинерционными термопарами и покрытые катализаторами. На поверхности таких катализаторов происходят процессы рекомбинации атомов и радикалов, сопровождающиеся значительным выделением тепла. Например, катализатор ZnO–Cr2О3 ускоряет реакцию рекомбинации атомов водорода. По повышению температуры зонда определяли концентрацию свободных частиц.
Итак, отличительными чертами цепных реакций являются следующие:
–большой квантовый выход при фотохимическом возбуждении;
–высокая чувствительность к присутствию примесей;
–зависимость скорости от формы и размера сосудов, в которых протекают реакции;
–наличие верхнего и нижнего пределов воспламенения.
Цепные реакции могут протекать с очень малыми и с большими скоростями.
Для неразветвленных цепных реакций, когда каждая активная частица дает начало одной цепи, скорость зависит от длины цепи ν и числа активных частиц n0, зарождающихся в единице объема в единицу времени:
U n0 .
Подобная реакция после периода индукции протекает спокойно. По мере уменьшения исходной концентрации скорость ее постепенно падает, т.к. согласно закону действующих масс уменьшение концентрации снижает число столкновений, а следовательно, величины n0 и ν.
Химическая кинетика. Лаб. практикум |
43 |

2.ЛАБОРАТОРНЫЕ РАБОТЫ
2.4.Цепные и фотохимические реакции
Однако во многих случаях каждая активная молекула-радикал порождает не один, а два и более новых радикала, так, что цепь разветвляется. Если такое разветвление цепей идет быстро, то реакция, в отличие от простой цепной, течет не стационарно, а самоускоряясь, достигая при высоких исходных концентрациях стадии самовоспламенения или взрыва. Изменение во времени скорости разветвленного цепного процесса описывается уравнением
U kn0 e 1 . |
|
Отсюда при не слишком малых значениях времени |
|
U kn e , |
(5) |
0 |
|
где φ – константа нарастания, характеризующая |
скорость разветвления; |
k – постоянная. |
|
В зависимости от значения n0 начальная скорость реакции может быть большей или меньшей. При достаточно малом n0 скорость близка к нулю, и реакция практически не регистрируется в течение длительного периода ин-
дукции |
|
t 1 , |
(6) |
доходящего до десятков минут. Затем скорость реакции быстро возрастает и, достигнув минимума, начинает падать вследствие расхода компонентов реакции.
Примером фотохимической реакции может быть разложение перекиси водорода, изучаемое в данной работе. Фотохимическое разложение H2O2 – это типично разветвленная цепная реакция с участием различных активных радикалов. Реакция начинается с фотохимического разложения Н2О2 на радикалы ОН• под действием ультрафиолетового излучения и протекает по следующему механизму:
1. |
Н2О2 + hν → 2OH• – реакция возникновения активных радикалов. |
||||
2. |
ОН• + Н2О2 |
|
→ НО2• + Н2О |
– реакции развития цепи. |
|
3. НО2• + (Н2О2)2 → Н2О4• + ОН• + Н2О |
|||||
4. |
Н2О4• + hν → 2ОН• + О2 |
– регенерация активных |
|||
5. |
Н2О4• + S → 2НО2• + S |
радикалов. |
|||
6. Н2О4• + S → Н2О2 + О2 + S |
– реакции гибели активных |
||||
7. |
ОН• + НО2• → Н2О + О2 |
||||
|
• |
+ НО2 |
• |
→Н2О2 + О2 |
радикалов. |
8. НО2 |
|
|
|||
Последняя реакция (восьмая) относится к цепным реакциям с «вырожденными разветвлениями». (Цепными реакциями с «вырожденными разветвлениями» называются такие, в которых в результате распада продуктов реакции образуются свободные радикалы. В данной реакции промежуточный продукт НО2- даст при взаимодействии с H2О2 радикал ОН• и молекулу тетраоксида водорода, приводящую далее к разветвлению цепи). В данном про-
Химическая кинетика. Лаб. практикум |
44 |

2.ЛАБОРАТОРНЫЕ РАБОТЫ
2.4.Цепные и фотохимические реакции
цессе разветвление осуществляется фотохимически за счет подвода энергии извне. Относительно устойчивым промежуточным продуктом является свободный радикал OH•.
Экспериментальнаячасть
Реактивы, приборы, посуда: кварцевая коническая или круглодонная колба объемом 250 мл, перманганат калия 0,01 н. раствор, раствор перекиси водорода (Н2О2), раствор фенола от 0,03 до 3 М, установка для изучения фотохимических реакций, секундомер.
Последовательность выполнения работы. Изучение кинетики фото-
химического разложения перекиси водорода в водном растворе проводится на установке, изображенной на рис. 5.
Раствор перекиси водорода в кварцевой колбе 1 облучают ультрафиолетовым светом от источника 2. Разлагаясь, Н2О2 выделяет кислород, объем которого измеряется с помощью градуированной бюретки 3, соединенной с уравнительным сосудом 4.
Рис. 5. Установка для изучения фотохимического разложения перекиси водорода
Перед работой следует промыть кварцевую колбу хромовой смесью и тщательно прополоскать дистиллированной водой. Затем налить в колбу, разведенную до определенной концентрации (по указанию преподавателя) перекись водорода. Концентрацию исследуемого раствора перекиси определяют титрованием 10 мл этого раствора 0,01 н. перманганатом калия. По количеству взятой перекиси рассчитывают предельный объем кислорода, который может выделиться при полном разложении.
Колбу располагают против отверстия защитного кожуха ртутнокварцевой лампы. Устанавливают на нуль уровень жидкости в измерительной бюретке, подняв при этом уравнительный сосуд до совпадения уровней. Необходимо удостовериться в герметичности установки. Затем включают лампу. Одновременно с зажиганием лампы пускают секундомер. Вначале отсчет объема кислорода производят каждые 2 мин. Когда скорость реакции
Химическая кинетика. Лаб. практикум |
45 |

2.ЛАБОРАТОРНЫЕ РАБОТЫ
2.4.Цепные и фотохимические реакции
возрастет – ежеминутно. Каждый отсчет осуществляют после выравнивания уровней бюретки и сосуда 4. Опыт прекращают, когда больше не происходит выделение газа. При отсчете объема газа требуется выравнивать уровни жидкости в уравнительном сосуде и газовой бюретке. После окончания опыта необходимо выключить лампу, вновь тщательно промыть кварцевую колбу.
Фотохимический распад перекиси водорода обладает повышенной чувствительностью к наличию в растворе примесей и состоянию стенок реакционного сосуда. Изучить скорость этого процесса можно, не только изменяя концентрацию взятой перекиси, но и проводя несколько последовательных опытов при одинаковой концентрации, добавляя для замедления скорости реакции до 3 мл 0,03 М раствора фенола. Скорость реакции зависит также от интенсивности облучения. Для изучения влияния этого параметра можно либо изменять расстояние между реакционной колбой и источником облучения, либо менять длительность облучения, проводя его лишь первые 15–30 мин. Фотохимический распад можно изучать также в зависимости от диаметра реакционного сосуда, используя колбы разного объема.
Экспериментальные данные заносят в табл. 9.
|
|
|
|
Таблица 9 |
|
|
|
|
|
|
|
Номер |
Время от начала |
Объем выделившегося |
Скорость реакции |
|
ln w |
измерения |
реакции τ, c |
кислорода VО2, мл |
w, мл/с |
|
|
|
|
|
|
|
|
По полученным данным строят кинетическую кривую в координатах VO2 = f(τ). Дифференцируя полученный график, находят значения скорости
реакции в каждый момент времени:
w vt2 vt1 . t2 t1
Построим графики зависимостей:
1.w = f(τ).
2.lg w = f(τ).
Логарифмическая форма уравнения (5) скорости цепной реакции является уравнением прямой:
lg w lg kn |
|
|
. |
(7) |
|
|
|
||||
0 |
|
2,303 |
|
|
|
|
|
|
|
||
В связи с убылью исходного вещества в ходе реакции имеет смысл обрабатывать по этому уравнению только данные, относящиеся к восходящей ветви кривой w = f(τ).
Построив график в координатах lg w–τ, по отрезку, отсекаемому на оси ординат, находят значение kn0, а по тангенсу угла наклона прямой – константу нарастания φ. Затем по соотношению (6) рассчитывают период индукции.
Химическая кинетика. Лаб. практикум |
46 |

2.ЛАБОРАТОРНЫЕ РАБОТЫ
2.4.Цепные и фотохимические реакции
Для подготовки к защите лабораторной работы можно воспользоваться следующей литературой: [1, с. 54–94; 4, с. 243–315; 12, с. 343–372, 418–467; 18, с. 373–387].
Контрольныевопросыизадания
1.Какие параметры оказывают влияние на скорость фотохимической, цепной реакции?
2.В чем отличие цепных реакций от каталитических, автокаталитических и простых сопряженных реакций?
3.Как можно сократить или удлинить период индукции?
4.Какое значение в практике имеют цепные фотохимические реакции?
5.Нарисуйте зависимость скорости цепной реакции от времени при условии, что количество центров бесконечно возрастает.
6.Сформулируйте основные законы фотохимии.
7.Дайте определение квантового выхода.
8.Опишите механизм Штерна – Фольмера.
2.5.Гетерогенныепроцессы
Лабораторная работа 6
Изучениекинетикитопохимическихреакций
Цельизадачиработы
Цели работы – исследование кинетики топохимической реакции разложения перманганата калия. В задачи работы входят построение кинетических кривых и нахождение эмпирических коэффициентов k и m уравнения Ерофеева.
Теоретическоевведение
Образование и диссоциация карбонатов, восстановление окислов и сульфидов, производство цемента, огнеупоров, керамики и многие другие химические превращения являются сложными гетерогенными процессами, имеющими огромное практическое значение. Их особенность состоит в том, что химическая стадия процесса сопровождается превращением в твердом состоянии, когда вещество с одной кристаллической решеткой исчезает и образуется продукт с другой структурой. Простейший пример такой реакции –
Химическая кинетика. Лаб. практикум |
47 |

2.ЛАБОРАТОРНЫЕ РАБОТЫ
2.5.Гетерогенные процессы
диссоциация Aтв Втв+ Сгаз . Указанные реакции называют топохимическими.
Для них характерно нарастание скорости в начальный период процесса и достижение ею предельного значения с последующим постепенным спадом до нуля (рис. 6, кривая 2). Одним из внешних признаков топохимической реакции служит S-образный вид кинетической кривой (рис. 6, кривая 1).
Топохимическая реакция начинается обычно на отдельных участках поверхности, в так называемых потенциальных центрах реакции, откуда она постепенно распространяется вглубь кристалла. Центры реакции – места на поверхности кристалла, где связи частиц в решетке ослаблены (вершины углов, ребра, дефекты на поверхности).
В первый период реакции образуются микроскопические зародыши ядер новой фазы (реакция идет с ничтожной скоростью). Образование зародышей ядер, в свою очередь, вызывает искажение материнской решетки, которое способствует возникновению новых зародышей. Одновременно увеличиваются и сами ядра, поверхность реакции растет, в результате повышается скорость химического взаимодействия. Этот период является автокаталитическим, т.к. образование продукта реакции, приводящее к увеличению поверхности раздела фаз, вызывает возрастание скорости. Скорость процесса будет максимальной при наибольшей величине поверхности реакции, когда межфазные границы отдельных зон, центров приходят в соприкосновение. После того как зоны перекрываются, поверхность начинает уменьшаться, и скорость реакции падает.
Рис. 6. Кинетика топохимической реакции: зависимости степени превращения (1) и скорости реакции (2) от времени процессса
Для того чтобы получить уравнение кинетики топохимической реакции, нужно знать или предположить, по крайней мере, следующие основные параметры:
–закон образования ядер новой фазы;
–закон роста ядер;
–форму ядер.
Возможны различные сочетания законов образования, роста и формы ядер продукта, поэтому существует большое количество уравнений, описывающих кинетику топохимических реакций. Но наиболее широко применяются лишь несколько из них.
Химическая кинетика. Лаб. практикум |
48 |

2.ЛАБОРАТОРНЫЕ РАБОТЫ
2.5.Гетерогенные процессы
На основании вероятности взаимодействия молекул данной системы и вне связи с предположениями об истинном механизме реакции Ерофеев получил уравнение
|
|
|
|
|
|
|
|
|
|
, |
(8) |
1 exp |
|
Pd |
|||
|
|
0 |
|
|
|
где – доля прореагировавших молекул к моменту времени ; P – вероятность того, что молекулы прореагируют в течение времени от до + d . Определив Р, можно получить кинетическое уравнение соответствующего процесса. В общем случае вероятность взаимодействия молекул в любой момент времени пропорциональна суммарной поверхности реакции, т.е. Р = А·S. Примем, что поверхность реакции изменяется во времени по степенному за-
кону S A' m |
. |
Тогда
P A" m , |
(9) |
где A, A', A'' – коэффициенты пропорциональности. Подставляя выражение (9) в выражение (8), получим
1 exp k m . |
(10) |
Здесь k, m – эмпирические коэффициенты, m = r + n, где r – число элементарных стадий при превращении зародыша в активно растущее ядро, а n зависит от числа направлений, в которых растут ядра (может быть равным 1, 2, 3). Коэффициенту k иногда приписывается смысл константы скорости. Величины k и m находят графически по экспериментальным данным. Для этого уравнение (10) представляют в линейной форме
|
1 |
|
|
|
ln k mln |
(11) |
|
ln |
|
|
|
||||
|
|
1 |
|
|
|
||
|
|
1 |
|
|
|
f (ln ) определяют k и m. |
|
и по графику в координатах ln |
1 |
|
|
|
|||
|
|
|
|
|
|
||
Химическая кинетика. Лаб. практикум |
49 |

2.ЛАБОРАТОРНЫЕ РАБОТЫ
2.5.Гетерогенные процессы
Экспериментальнаячасть
Реактивы, приборы, посуда: исследуемое вещество – перманганат калия (KMnO4), экспериментальная установка для изучения топохимических реакций.
Последовательность выполнения работы. Схема экспериментальной установки для изучения кинетики топохимических реакций с выделением газа изображена на рис. 7.
Она состоит из печи сопротивления 1, температура в которой регулируется лабораторным автотрансформатором 2, термометра 3 для измерения температуры в печи, пробирки 4 с исследуемым веществом, газометрической бюретки 5 с уравнительным сосудом 6 для измерения объема выделяющегося при реакции газа.
В работе нужно снять кинетические кривые термического разложения исследуемого вещества при двух температурах (задает преподаватель).
Порядок проведения всех измерений одинаков, и они производятся с равными навесками вещества.
Автотрансформатор включают в сеть переменного тока и согласно градуировочной кривой выставляют нужное напряжение, соответствующее нижней температуре опыта. Пробирку и термометр располагают рядом по центру шахты печи так, чтобы нижний срез находился примерно в середине зоны нагревателя. После того как температура установится постоянной на заданной величине и будет неизменной в течение 10 мин, в пробирку засы-
пают навеску исследуемого вещества ( 0,3 г), плотно закрывают пробкой на отводном шланге, включают секундомер и отмечают первое показание на бюретке.
Рис. 7. Схема экспериментальной установки
Время между отсчетами следует выбирать, исходя из необходимости построения точной кинетической кривой. Как правило, в начале и конце
реакции отрезки времени более продолжительны и с повышением темпе-
Химическая кинетика. Лаб. практикум |
50 |
2.ЛАБОРАТОРНЫЕ РАБОТЫ
2.5.Гетерогенные процессы
ратуры уменьшаются. Наиболее целесообразно производить отсчеты времени, в течение которого объем выделяющегося газа изменится на определенную величину (1–2 мл). Окончанием реакции считают время, когда объем газа в бюретке в течение 10–15 мин остается неизменным.
После окончания опыта исследуемое вещество извлекают из реакцион-
ной пробирки и вновь взвешивают для определения потери веса gо.п в результате реакции. Полученное значение сравнивают с весом выделившегося
газа и теоретическим значением gт, рассчитанным по стехиометрическому уравнению, предполагая, что реакция идет до конца. Если в качестве исследуемого вещества взят перманганат калия KМnO4, то реакция его разложения в суммарном виде может быть выражена уравнением, справедливым для температур вблизи 200 оС:
2KMnO4 K2MnO4 MnO2 O2
Экспериментальные данные для каждой температуры представляют в
виде табл. 10 (V – объем газа в данный момент времени; V – конечный объем выделившегося газа).
Таблица 10
Навеска g = … г, t оC = …, gо.п = …, gт = …
№ |
|
V , мл |
= V /V |
lnln [1/(1 – )] |
ln |
k |
m |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Кроме того, необходимо построить следующие графики:
1.Зависимость V = f( ).
2.Кинетические кривые f ( ) .
3.Зависимости скорости реакции от времени.
4.Графики для определения k и m по уравнению Ерофеева.
Для подготовки к защите лабораторной работы можно воспользоваться следующей литературой: [11, с. 177–222; 18, с. 396–408].
Контрольныевопросыизадания
1. Проанализируйте обобщенную кинетическую кривую f ( ) : начальный период, периоды индукции, ускорения и спада.
2.Запишите линейный и экспоненциальный законы зародышеобразования в случае, когда превращение потенциального центра в зародыш происходит в одну стадию.
3.Запишите уравнение Праута – Томпкинса.
4.В чем заключается суть концепции зародышеобразования по разветвленному цепному механизму?
5.Запишите уравнение Ерофеева и покажите, как графически определить постоянные в этом уравнении.
6.В чем заключаются особенности топохимических реакций?
7.Приведите примеры топохимических реакций.
Химическая кинетика. Лаб. практикум |
51 |

2.ЛАБОРАТОРНЫЕ РАБОТЫ
2.5.Гетерогенные процессы
Лабораторная работа 7
Изучениекинетикииспаренияжидкости
идиффузииеепаровввоздухе
Цельизадачиработы
Цель работы – изучение процессов стационарного и нестационарного испарения жидкости. В задачи работы входит определение коэффициента диффузии летучего компонента в стационарном и нестационарном режимах испарения, а также скорости нестационарного испарения.
Теоретическоевведение
Всякий гетерогенный процесс включает в себя несколько стадий. Основные из них транспорт реагирующих веществ к реакционной поверхности, собственно химическая реакция, отвод продуктов реакции в объем. В зависимости от условий проведения процесса и его особенностей наиболее медленной может быть любая из трех стадий. Если лимитирующими являются первая и третья стадии, то скорость процесса зависит от скорости переноса вещества посредством диффузии. Такой режим называют диффузионным.
Большое значение имеют диффузионные влияния в процессах испарения жидкостей и сублимации твердых тел. Они играют ведущую роль при сушке, перегонке и т.д. – везде, где скорость процесса лимитируется отводом пара от поверхности конденсированной фазы. Процесс диффузии подчиняется первому закону Фика:
Y D dC |
|
, |
(12) |
d x |
|
|
|
где Y 1s ddm – количество вещества m, проходящее за время d через сече-
ние S (диффузионный поток, г/см2 с); D – коэффициент диффузии; ddCx – гра-
диент концентрации.
Знак «–» показывает, что диффузия идет в направлении убывания концентрации. Величина D представляет собой удельную скорость диффузии, т.е. количество вещества, проходящего в единицу времени через единицу поверхности при единичном градиенте концентрации. Как следует из выражения (12), коэффициент D имеет размерность D = (Длина)2/Время, а выражают его обычно в см2/с.
Кинетическая сторона диффузии ясна из следующих рассуждений. Если на границе раздела фаз концентрация испаряющегося вещества будет Сs,
Химическая кинетика. Лаб. практикум |
52 |

2.ЛАБОРАТОРНЫЕ РАБОТЫ
2.5.Гетерогенные процессы
ана расстоянии – С и равна концентрации в глубине объема V, то можно приближенно заменить
|
dC |
|
|
|
Cs C |
. |
(13) |
||
d x |
|
|
|||||||
|
|
|
|
|
|||||
Тогда вместо выражения (12) имеем |
|
Cs C |
|
|
|||||
Y D |
. |
(14) |
|||||||
|
|
|
|||||||
Разделив обе части уравнения (14) на объем и переходя к концентрации, получим
Y |
|
1 d m 1 |
|
dC D |
|
C |
C |
|
||
V |
|
|
d |
|
|
d V |
|
|
s |
. |
V S |
S |
|
||||||||
Отсюда скорость диффузии
dC |
|
D S C C . |
(15) |
d |
|
V s |
|
Таким образом, скорость диффузии кинетически подчиняется уравнению первого порядка относительно концентрации в объеме. Интегрирование выражения (15) дает обычное выражение для константы массопереноса
VDS , которая является аналогом константы скорости
1 ln Cs C0 ,
Cs C
где С0 – начальная концентрация диффундирующего вещества в объеме. Диффузия – процесс относительно медленный. Это объясняется тем,
что каждая молекула диффундирующего вещества испытывает огромное число соударений и перемещается по сложной траектории, длина которой несоизмеримо велика по сравнению с расстоянием, проходимым молекулой в направлении диффузии.
Вследствие этого лишь небольшая часть молекул, вырвавшихся из жидкости, успевает удалиться от ее поверхности на значительное расстояние. В тонком слое газа над самой поверхностью накапливаются молекулы испаряющегося вещества, и парциальное давление паров растет до тех пор, пока не станет почти равным давлению насыщенного пара. В общем случае говорят: если процесс лимитируется диффузией, то около поверхности раздела фаз устанавливается состояние, близкое к равновесному.
Для экспериментального определения коэффициентов диффузии чаще всего создают такие условия, в которых процесс испарения и диффузии паров протекает стационарно. В стационарном процессе скорость его, а также концентрация вещества в любой точке системы не меняются со временем. Такой процесс диффузии легко рассчитать.
Химическая кинетика. Лаб. практикум |
53 |

2.ЛАБОРАТОРНЫЕ РАБОТЫ
2.5.Гетерогенные процессы
Например, для описания скорости стационарного испарения жидкости в вертикальной цилиндрической трубе, у верхнего среза которой поддерживается постоянное парциальное давление паров Р0, Стефан получил уравнение
YC D ln P P0 ,
P Ps
позволяющее выразить коэффициент диффузии через измеряемые в опыте величины:
D |
h h d RT |
|
, |
(16) |
|
|
|
|
|||
|
P P0 |
|
|
||
|
M P ln |
|
|
||
|
P P |
|
|||
|
|
|
|
||
|
s |
|
|||
где h – расстояние от поверхности жидкости до верхнего среза трубы; h – измерение высоты уровня жидкости в результате испарения за время ;
d MRTP – плотность диффундирующего вещества, г/см3, соответствующая
его парциальному давлению, которое равно общему атмосферному давлению Р; R – универсальная газовая постоянная; М – молекулярный вес жидкости; Рs – давление насыщенных паров жидкости при температуре Т.
С повышением температуры коэффициент диффузии возрастает экспоненциально в жидкостях и твердых телах:
DD0ехр RЕT
ипо степенной зависимости в газах:
D2 |
|
|
n |
T2 |
. |
||
D1 |
|
T1 |
|
Значение показателя степени в последнем уравнении лежит обычно в пределах 1,6–2,0.
Анализ нестационарного процесса испарения жидкости из вертикальной цилиндрической трубы показывает, что данный процесс должен подчиняться уравнению
x |
2 |
2D |
C0 |
, |
(17) |
|
1 |
||||
|
|
|
C20 |
|
|
где х – расстояние, на которое переместилась в трубке граница жидкость–пар за время ; С10 – концентрация насыщенного пара жидкости на границе,
кг/м3; С20 – концентрация воздуха на межфазной границе, кг/м3; C C10 C20 .
Химическая кинетика. Лаб. практикум |
54 |

2.ЛАБОРАТОРНЫЕ РАБОТЫ
2.5.Гетерогенные процессы
Экспериментальнаячасть
Реактивы, приборы, посуда: ацетон, пробирки, штатив с держателем, катетометр.
Последовательность выполнения работы. В держатели штатива за-
жимают две тонкие (4–6 мм диаметром) стеклянные пробирки. Одна из них служит для определения коэффициентов диффузии пара жидкости при стационарном процессе испарения по уравнению Стефана (16) (метод 1), другая – для расчета скорости нестационарного испарения по уравнению (17) (метод 2).
Метод 1. Исследуемую жидкость наливают в пробирку на 50–80 мм ниже ее верхнего среза. Через 15–20 мин, в течение которых происходит установление стационарного состояния, начинают измерения при помощи катетометра (см. прил. 4). Измерять h нужно с большой точностью, т.к. испарение идет медленно и h выражается десятыми долями миллиметра. Отсчеты снимают через 5 мин. За время опыта снимают 6-7 отсчетов и строят график h f ( ) , позволяющий судить о стационарности процесса испарения
d h |
|
. По уравнению (16) рассчитывают коэффициент диффузии пара |
|
const |
|
d |
|
|
жидкости в воздухе. Поскольку в данной работе сложно определить Р0, его значение принимают равным нулю, хотя это можно делать только в случае полного удаления паров у среза трубки, что выполняется, например, при измерении коэффициентов диффузии паров методом увлечения. За высоту h принимают среднее ее значение за время опыта (высота диффузионного пространства):
h h0 2h .
Измерения заносят в таблицу и представляют в виде графиков:
1.h f ( ).
2.h f ( ) .
Метод 2. Пробирку заполняют исследуемой жидкостью полностью до верхнего среза. Измерения перемещения межфазной границы во времени производят микрометром сразу после заполнения с промежутками между измерениями 2 мин. Всего снимают 6–7 отсчетов.
Измерения заносят в таблицу и представляют в виде графиков:
1.х = f( ).
2.х2 = f( ).
3.Зависимость скорости испарения I id от (i – скорость перемеще-
ния межфазной границы, i d x d ).
Химическая кинетика. Лаб. практикум |
55 |

2.ЛАБОРАТОРНЫЕ РАБОТЫ
2.5.Гетерогенные процессы
По тангенсу угла наклона второго графика с учетом давления насыщенного пара исследуемой жидкости при температуре опыта рассчитывают D и сравниваютсполученнымзначениемпометоду1 илитературнымиданными.
Концентрации С10 и С20 на межфазной границе находят по формуле Ci Nid (Ni – мольная доля газа, равная отношению парциального давления
газа к общему давлению), используя взятое из справочника значение давления насыщенного пара жидкости при температуре опыта и рассчитывая плотность пара жидкости dп, считая его идеальным газом:
dп MRTP .
Для подготовки к защите лабораторной работы можно воспользоваться следующей литературой: [4, с. 390–391; 18, с. 387–396].
Контрольныевопросыизадания
1.Как зависит скорость испарения жидкости от температуры?
2.По каким признакам можно судить о том, в каком режиме идет процесс: в кинетическом или диффузионном?
3.Дайте определение диффузии. Каков физический смысл коэффициента диффузии?
4.Что означает выражение «стационарный режим процесса»?
5.Запишите уравнение Стефана.
6.Покажите, как графически определить коэффициент диффузии паров жидкости в нестационарном процессе испарения.
7.Как графически определить энергию активации процесса диффузии?
Лабораторная работа 8
Изучениекинетикирастворенияидиффузии вводныхрастворах
Цельизадачиработы
Цель работы – изучение кинетики растворения твердых веществ в водных растворах. В ходе выполнения работы должны быть решены следующие задачи: 1) ознакомление с методом вращающегося диска; 2) определение скорости растворения в зависимости от скорости вращения диска и от концентрации раствора; 3) расчет коэффициента диффузии.
Химическая кинетика. Лаб. практикум |
56 |

2.ЛАБОРАТОРНЫЕ РАБОТЫ
2.5.Гетерогенные процессы
Теоретическоевведение
Многие гетерогенные реакции идут в диффузионном режиме. Это особенно относится к высокотемпературным реакциям, имеющим промышленное значение. Как правило, скорости таких процессов в неподвижной среде очень малы, и поэтому их стараются повысить, прибегая к размешиванию при помощи специальных приспособлений.
Перенос вещества в движущейся жидкости обусловлен двумя различными механизмами. Во-первых, при наличии разности концентраций в жидкости возникает молекулярная диффузия в соответствии с законом Фика, вовторых, частицы вещества, растворенного в жидкости, увлекаются последней в процессе ее движения и переносятся вместе с ней. Совокупность обоих процессов называют конвективной диффузией вещества в жидкости или газе.
Исследование диффузионной кинетики растворения твердых тел в жидкости привело к установлению соотношения количества вещества, растворившегося в единицу времени, аналогичного уравнению (14) из работы 7. Вследствие разности концентраций растворяющегося вещества около по-
верхности Сs и в глубине объема возникает диффузионный слой толщиной , являющийся основным препятствием для частиц на пути к границе раздела. По теории Нернста, распределение концентрации внутри этого слоя носит линейный характер. В слое жидкость считается неподвижной, за пределами этого слоя она находится в движении, т.е. перемешивается, что ведет к установлению постоянной концентрации в объеме. Теория не позволяет ни вычислить , ни оценить значение потока Y, т.е. это качественная теория диффузионной кинетики гетерогенных взаимодействий.
Позднее было доказано, что жидкость в пределах диффузионного слоя не неподвижна и распределение концентрации в нем не описывается линейным законом. Это значит, что уравнение (14), приведенное в работе 7, не позволяет точно вычислить скорость массопереноса. Для этого необходимо знать скорость и направление движения жидкости в пределах диффузионного слоя, а также их влияние на диффузионный перенос вещества.
Наиболее простой вид имеет уравнение конвективной диффузии для реакционной поверхности в виде вращающегося диска. Анализ дает следующую картину движения жидкости около диска. При вращении диска в вязкой среде слой жидкости, непосредственно прилегающий к поверхности, вращается вместе с ней. Наличие вязких сил вызывает вращение и более удаленных слоев, однако с удалением от поверхности вращательное движение ослабевает. Под действием центробежных сил вращающиеся слои приобретают радиальную скорость и отбрасываются к периферии. На их место подходят новые порции жидкости из глубины объема.
Толщина слоя жидкости, увлекаемого во вращение (так называемого гидродинамического слоя 0), оказывается пропорциональной квадратному корню из соотношения кинематической вязкости среды к угловой скорости вращения диска 2 n :
Химическая кинетика. Лаб. практикум |
57 |

2.ЛАБОРАТОРНЫЕ РАБОТЫ
2.5.Гетерогенные процессы
0 3,6 ,
где n – скорость вращения диска, об/с. Скорость W, с которой жидкость подходит к поверхности диска, определяется выражением
W 0,836
и не зависит от расстояния до оси диска.
Далее анализ решений уравнения конвективной диффузии показывает, что при вращении диска всю жидкость можно разбить на две области: область постоянной концентрации во всем объеме вдали от поверхностной реакции и область быстрого изменения концентрации непосредственно вблизи этой поверхности. Последняя область представляет собой тонкий слой жидкости, в котором проявляется молекулярная диффузия. Поэтому этот слой называют диффузионным пограничным слоем . Расчеты показывают, что в пределах слоя концентрация раствора изменяется быстро и почти линейно с расстоянием. Тогда выражение для диффузионного потока можно представить приближенно в линейном виде:
Y D |
Cs C |
, |
(18) |
|
т.е. в таком виде, как и в теории Нернста. Однако в данном случае является определенной функцией свойств жидкости и скорости ее движения, а также коэффициента диффузии:
1 |
1 |
1 |
(19) |
1,61 D2 |
6 |
2 . |
Размеры диффузионного и гидродинамического 0 слоев связаны соотношением
1
0,45 D 2 0 .
Из этого выражения следует, что для жидких сред толщина слоя составляет всего несколько процентов от 0, т.к. D << ν. В то же время в газах, где D ν, они сравнимы по величине.
Толщина диффузионного слоя, как это следует из уравнения (19), одинакова по всей поверхности диска, что ведет к равнодоступности ее в диффузионном отношении. Наличие свойства равнодоступности выгодно выделяет поверхность вращающегося диска среди других реакционных поверхностей. Например, толщина диффузионного и гидродинамического слоев на плоской поверхности, которая смывается струей раствора, зависит от расстояния до ее переднего края, что приводит к неравномерному растворению или осаждению вещества на поверхности пластины.
Химическая кинетика. Лаб. практикум |
58 |
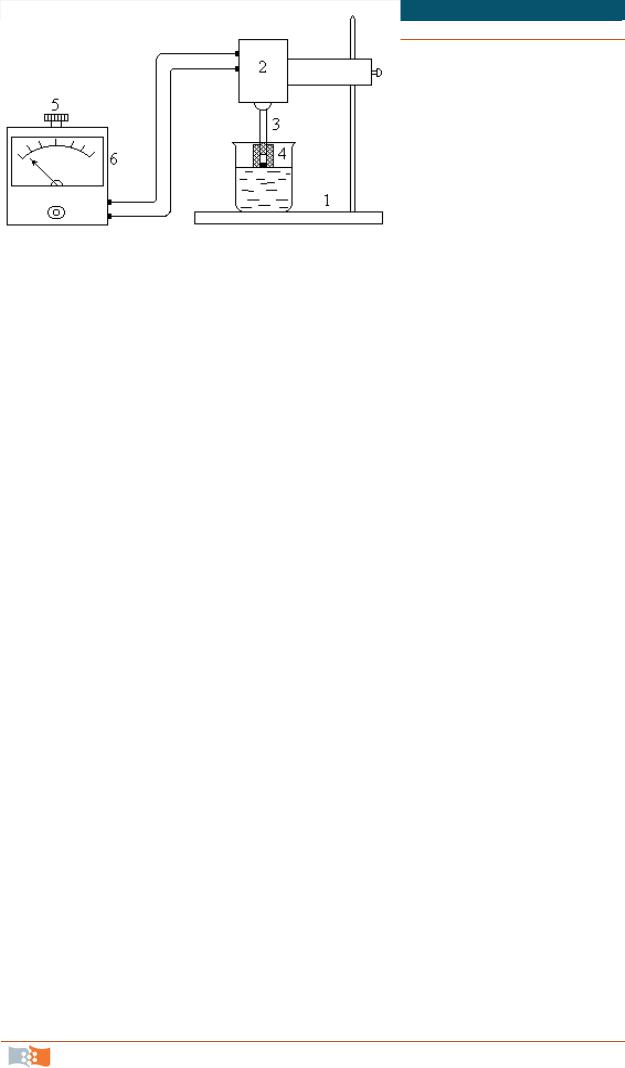
2.ЛАБОРАТОРНЫЕ РАБОТЫ
2.5.Гетерогенные процессы
Зная величину , можно по уравнению (18) рассчитать поток диффундирующих частиц к поверхности диска, т.е. скорость гетерогенной реакции на диске в диффузионном режиме Y , г/(см3·с):
2 |
- |
1 |
1 |
Cs C . |
|
Y 0,62D3 |
6 |
2 |
(20) |
На основании экспериментальных данных о величинах Y и , а также по взятым из справочника величинам Cs и можно определить коэффициент диффузии вещества. Находя зависимость скорости взаимодействия от температуры, можно вычислить энергии активации скорости процесса Е и диффу-
зии ЕD.
Приведенный анализ относится к ламинарному (или безвихревому) течению жидкости вблизи вращающегося диска.
Экспериментальнаячасть
Реактивы, приборы, посуда: металлические образцы или соли, прессованные в виде таблеток, водные растворы кислот или растворы солей изучаемых металлов, секундомер, установки для изучения кинетики гетерогенных взаимодействий методом вращающегося диска, аналитические весы, наждачная бумага.
Последовательность выполнения работы. Целью данной работы яв-
ляется изучение кинетики растворения твердых веществ в растворах. Определение скорости растворения проводят на установке, схема которой представлена на рис. 8.
Рис. 8. Схема установки для изучения кинетики гетерогенных взаимодействий методом вращающегося диска
Установка состоит из станины 1 с вертикальным стержнем, по которому перемещается электродвигатель 2. Электродвигатель приводит во вращение шток 3, на конце которого закреплена обойма с образцом исследуемого вещест-
Химическая кинетика. Лаб. практикум |
59 |

2.ЛАБОРАТОРНЫЕ РАБОТЫ
2.5.Гетерогенные процессы
ва 4. Скорость вращения образца можно плавно изменять от 0 до 1 500 об/мин регулировочным устройством 5 и регистрировать при помощи стрелочного индикатора 6. Для перевода показаний индикатора в значения скорости, об/мин, используют градуировочную кривую.
В работе изучают кинетику растворения металлических или прессованных из солей образцов (выдает преподаватель) в водных растворах кислот либо соответствующих солей, концентрацию которых тоже задает преподаватель. Перед проведением опыта образцы, взвешенные на аналитических весах с точностью до 0,0001 г, вставляют в обойму из алюминия или фторопласта и обрабатывают наждачной бумагой с целью выравнивания торцов образца и обоймы и получения гладкой поверхности. После этого обойму навинчивают на шток, включают электродвигатель, по индикатору устанавливают одно из заданных значений скорости вращения и погружают образец в раствор на 2-3 мм. Одновременно с погружением включают секундомер. Время растворения (обычно 5–15 мин) выбирают с таким расчетом, чтобы убыль веса образца составляла не менее 0,03 г, т.к. малые значения g вызывают большие относительные погрешности при взвешивании и дальнейшем расчете Y и D. После опыта достают образец из обоймы, просушивают и повторно взвешивают. Так проводят 3-4 опыта при различных скоростях вращения, но одинаковых значениях времени и концентрации раствора.
По данным опытов рассчитывают скорости растворения Y, используя формулу
Y Sg ,
где S – площадь рабочей поверхности образца.
Значение коэффициента диффузии D вычисляют по формуле (20). Результаты измерений и расчетов заносят в табл. 11.
Далее изучают кинетику растворения в зависимости от концентрации раствора. Для этого проводят 3-4 опыта при одном и том же значении скорости вращения диска.
На основании результатов опытов строят графики:
1)Y f ( ) ;
2)Y f (C) ;
3)lgY f (lgC) .
Таблица 11
= … см2/с, t = … оC, исследуемое вещество …, Сs = … г/см3
Химическая кинетика. Лаб. практикум |
60 |
2.ЛАБОРАТОРНЫЕ РАБОТЫ
2.5.Гетерогенные процессы
№ |
n, об/мин |
|
, с |
g |
, г |
g |
, г |
|
g, г |
Y, г/см3с |
, с |
-0,5 |
С, г/см3 |
D, см2/с |
|
|
|
1 |
|
2 |
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
После чего проводят расчет поверхностных концентраций Сs. Считают, что если процесс растворения протекает в диффузионном ре-
жиме, то около поверхности раздела твердое – жидкость устанавливается концентрация реагентов, близкая к равновесной, т.е. соответствующая предельной растворимости данного вещества в растворителе при температуре опыта (определяют по справочнику).
Исходя из значения растворимости, рассчитывают весовые доли растворяемого вещества и растворителя в приповерхностном слое раствора – N1 и N2 (весовая доля – отношение массы данного компонента к массе системы (раствора)). Принимая, что плотности в этом слое, кг/см3, складываются аддитивно, с учетом весовых долей и значения плотности твердого вещества d находят концентрацию Сs : Cs = Nid. (Если расчет концентраций вызывает трудности, можно обратиться к прил. 5).
Для подготовки к защите лабораторной работы можно воспользоваться следующей литературой: [1, с. 224–238; 4, с. 390–391; 18, с. 387–396].
Контрольныевопросыизадания
1.В чем заключается отличие молекулярной и конвективной диффузии?
2.Какой параметр вводится теорией конвективной диффузии Нернста и от чего он зависит?
3.Как связаны константы скоростей химической реакции и диффузии в случае, когда гетерогенный процесс идет в переходной области?
4.Почему при высоких температурах лимитирующей стадией гетерогенного процесса является диффузия?
5.Перечислите отличительные признаки протекания процесса во внешнедиффузионной области.
6.В чем заключается отличие внутренней диффузии от внешней?
7.Какая существует количественная связь между опытной энергией активации во внутридиффузионной области и энергией активации во внутренней кинетической области?
Химическая кинетика. Лаб. практикум |
61 |

2.ЛАБОРАТОРНЫЕ РАБОТЫ
2.5.Гетерогенные процессы
Лабораторная работа 9
Изучениекинетикипроцессовтравления полупроводников
Цельизадачиработы
Цельработы– изучениекинетикивзаимодействиятвердоетело– жидкость на примере процесса травления полупроводников. В задачи работы входит определение скорости реакции методом графического дифференцирования и глубины нарушенного слоя.
Теоретическоевведение
Травление – специальная обработка поверхности твердых тел с целью:
– очистки поверхности кристалла, придания ей необходимого рельефа, удаления поверхностного слоя после механической обработки. Травление применяется при изготовлении любых полупроводниковых приборов. Атомы примесей и загрязнений, находящиеся на поверхности, могут при процессе термообработки диффундировать в объем кристалла, незаметно и необратимо изменять его свойства. Кроме того, сами поверхностные загрязнения (окислы, металлические ионы, адсорбированные примеси пара воды, дефекты структуры поверхностного слоя) оказывают огромное влияние на электрофизические свойства полупроводников;
– подготовки поверхности для металлографического исследования. При этом в результате травления увеличивается оптический контраст между разнородными участками поверхности. Исследование результатов под микроскопом (металлографический метод) дает возможность определить кристаллическую структуру слитка, его фазовый состав, степень однородности.
Травление рассматривают как многостадийный процесс, в него включаются следующие этапы:
1)диффузия реагента к поверхности;
2)адсорбция реагента;
3)поверхностная реакция;
4)десорбция продуктов взаимодействия;
5)диффузия продуктов реакции от поверхности.
Скорость суммарного процесса зависит от наиболее медленной стадии (контролирующей). При умеренных температурах скорость травления определяется сдачей химического взаимодействия, реже – процессом диффузии. При высоких температурах контролирующей стадией служит диффузия. Адсорбция и десорбция характеризуются малыми энергиями активации, проте-
Химическая кинетика. Лаб. практикум |
62 |

2.ЛАБОРАТОРНЫЕ РАБОТЫ
2.5.Гетерогенные процессы
кают (сравнительно с другими этапами) быстро и потому лишь изредка лимитируют процесс.
Поверхностная реакция. Скорость взаимодействия между атомами кристалла и частицами травителя должна быть пропорциональна концентрации атомов кристалла и частиц травителя. Концентрация атомов зависит от природы материала и является постоянной величиной для данного полупроводника. Поэтому скорость химической реакции может быть определена из соотношения
v kCse |
Ea |
RT , |
(21) |
|
где k – единичная скорость реакции; Сs – концентрация травителя в поверх-
ностном слое; ke Ea RT – константа скорости реакции; Еа – энергия активации реакции; R – универсальная газовая постоянная; Т – абсолютная температура.
Из формулы (21) следует, что скорость химической реакции зависит от природы реагентов, концентрации, температуры и энергии активации. При прочих равных условиях температурная зависимость скорости реакции тем больше, чем выше Еа. Незначительное снижение Еа приводит к большим увеличениям скорости. Энергия активации зависит от структуры и состояния поверхности материала. Минимальной Еа будут обладать участки с максимальным количеством разорванных или ослабленных связей, т.е. участки, которые имеют отрицательные значения электродного потенциала. На грубо обработанных шероховатых поверхностях травление протекает с меньшей энергией активации. Так как различные кристаллографические плоскости структуры полупроводника имеют разное значение Еа, то скорость травления зависит от ориентации пластин. Посторонние атомы, дислокации и другие дефекты структуры могут повышать или понижать энергию активации растворения.
Если самой медленной стадией процесса считается химическая реакция, то травители называются селективными. При селективном травлении выявляются неоднородные поверхности.
Диффузия. Согласно теории Нернста, диффузионный поток определя-
ется выражением |
|
|
||
Y |
D C Cs |
Kдиф C Cs , |
(22) |
|
|
||||
|
|
|
||
где D – коэффициент диффузии; С, Сs – концентрации вещества в объеме и в поверхностном слое; – толщина диффузионного слоя; Kдиф – единичная скорость процесса диффузии.
В начальный момент времени травления С = Сs, и диффузия не происходит, условия для протекания химической реакции наиболее благоприятны. Травление селективное. Через некоторое время концентрация травителя
Химическая кинетика. Лаб. практикум |
63 |

2.ЛАБОРАТОРНЫЕ РАБОТЫ
2.5.Гетерогенные процессы
вблизи поверхности снизится до значения Сs, скорость диффузии возрастет, а скорость реакции, наоборот, уменьшится. При достижении стационарного состояния обе скорости станут равными по величине. С этого момента Сs и скорость травления больше изменяться не будут.
Диффузионный контроль наблюдается для растворов травителей с низкими концентрациями реагентов, при высоких температурах или при процессах, сопровождающихся образованием плохо растворимых продуктов. При этом скорость процесса зависит от интенсивности перемешивания, вязкости раствора и слабо зависит от температуры и структуры поверхности. Незначительное влияние температуры на скорость диффузии связано с небольшой энергией активации диффузии, составляющей всего несколько килоджоулей на моль, в то время как энергия активации химических реакций имеет порядок десятков килоджоулей на моль. Травители с диффузионным контролем называются полирующими. Особенность их действия заключается в сглаживании шероховатостей, нечувствительности к физическим и химическим неоднородностям поверхности.
Из уравнения (21) и уравнения (22) находим выражение для Сs и подставляем его в (21):
v Kдиф ke Ea RT C . Kдиф ke Ea RT
При условии Kдиф > kе-Еа/RT травление будет селективным, скорость процесса определяется микроструктурой данного участка (его Еа):
vтр ke Ea RT С.
При условии Кдиф < kе-Еa/RT травление полирующее, скорость процесса практически не зависит от энергии активации:
vтр Kдиф С .
При механической обработке поверхности полупроводниковых материалов образуются нарушенные слои, отличающиеся по своей структуре от глубинных слоев монокристалла. Толщину нарушенного слоя можно оценить по изменению скорости травления при стадийном растворении механически обработанных образцов, поскольку механически обработанная поверхность травится быстрее, чем совершенный монокристалл.
Постоянство скорости травления указывает на полное снятие нарушенного слоя. Скорость травления определяется по уменьшению толщины об-
Химическая кинетика. Лаб. практикум |
64 |

2.ЛАБОРАТОРНЫЕ РАБОТЫ
2.5.Гетерогенные процессы
разца в единицу времени. Процесс определения глубины нарушенного слоя состоит из следующих этапов:
1)очистка поверхности образца;
2)взвешивание образца перед травлением;
3)удаление с поверхности в полирующем травителе тонкого слоя ма-
териала;
4)определение количества удаленного полупроводникового материала (повторное взвешивание).
Этапы 2–4 повторяются до полного удаления нарушенного слоя (постоянство скорости травления).
Экспериментальнаячасть
Реактивы, приборы, посуда: кремниевые пластины, органический растворитель (толуол, изопропиловый спирт, ацетон, четыреххлористый углерод), травители СР-8 или Уайт, аналитические весы, фторопластовый стакан, пинцет с фторопластовым наконечником.
Последовательность выполнения работы. Поверхность кремниевой пластины перед травлением обезжиривают в органическом растворителе. Обезжиривание проводится кипячением пластины в органическом растворителе или тщательной протиркой ватным тампоном, смоченным в растворителе. Затем образец взвешивают на аналитических весах с точностью до 10-4 г.
Для определения глубины нарушенного слоя на кремнии используются следующие травители: СР-8 (НNO3:HF = 3:1) и Уайт (HNO3:HF = 2:1). Оба травителя являются полирующими (при использовании травителей, содержащих плавиковую кислоту, необходима фторопластовая посуда).
Пластины кремния закрепляют во фторопластовый держатель (можно использовать пинцет с фторопластовыми наконечниками) и травят на каждом этапе по 20–30 с при комнатной температуре. После травления образцы промывают дистиллированной водой последовательно в трех сосудах. После каждого цикла травления определяют изменение массы пластины gi. Процесс
повторяют до тех пор, пока изменение массы не станет постоянным. После полного удаления нарушенного слоя строят график зависимости количества удаленного материала gi от общего времени травления. Поскольку скорость травления пропорциональна количеству удаленного материала в единицу времени, то график gi
= f( ) характеризует изменение скорости в
процессе травления.
Рис. 9. Кривая gi = f( )
Химическая кинетика. Лаб. практикум |
65 |

2.ЛАБОРАТОРНЫЕ РАБОТЫ
2.5.Гетерогенные процессы
Кривая gi = f( ) имеет два участка: спада и постоянства скорости травления (рис. 9). Зная точку перегиба, можно рассчитать глубину нарушен-
ного слоя , мкм, по формуле
n
2 gi
|
1 |
104 , |
|
Sd |
|||
|
|
где 2 gi – общее количество удаленного материала до точки перегиба на кривой, г; S – площадь поверхности образца, см2; d – плотность исследуемого материала, г/см3.
Строят график gi = f( ), по нему методом графического дифференцирования находят скорость реакции в различные моменты времени. Результаты измерений и расчетов заносят в табл. 12.
Таблица 12
№ |
, с |
g1, г |
g2, г |
gi, г |
gi, г |
, мкм |
i |
Состав травителя, |
|
концентрация |
|||||||||
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
Для подготовки к защите лабораторной работы можно воспользоваться следующей литературой: [5, 22].
Контрольныевопросыизадания
1.Какие факторы и как влияют на Kдиф и k?
2.Какая стадия является лимитирующей при полирующем и селектив-
ном травлении?
3.Как зависит от температуры скорость реакции при селективном и полирующем травлении?
4.Каков порядок реакции при травлении кремния в полирующем тра-
вителе?
5.В чем заключается причина образования ямок травления?
6.Назовите отличия полирующего травления от селективного.
7.Как скорость процесса с участием твердой фазы связана со скоростями отдельных стадий?
Химическая кинетика. Лаб. практикум |
66 |

2.ЛАБОРАТОРНЫЕ РАБОТЫ
2.6.Основныеположениякатализа
Лабораторная работа 10
Изучениескоростиреакциийодированияацетона
Цельизадачиработы
Цель работы – исследование кинетики автокаталитической реакции на примере реакции йодирования ацетона. В задачи работы входят ознакомление с работой спектрофотометра «SPEKOL-1300», расчет константы скорости реакции йодирования, изучение первичного и вторичного солевых эффектов.
Теоретическоевведение
Йодирование ацетона катализируется в водных растворах протонами
CH3 CO CH3 I2 H CH3 CO CH2I H I
ипротекает в две стадии:
1.Энолизация ацетона (с образованием промежуточного вещества):
CH3 C CH3 H CH3 Ñ CH3 CH3 C CH2 H
|| |
| |
| |
O |
OH |
OH |
2. Взаимодействие йода с энольной формой:
CH3 C CH2 I2 CH3 С CH2I I H
| |
|| |
OН |
OH |
Первая реакция протекает значительно медленнее второй, поэтому скорость процесса определяется скоростью первой стадии. Скорость реакции йодирования ацетона зависит от концентрации ацетона и ионов водорода, но не йода. В результате реакция проходит по второму порядку. Реакция осуществляется автокаталитически, т.к. ускоряется одним из продуктов реакции (Н+). В нейтральном разбавленном водном растворе она протекает очень медленно.
Скорость расходования ацетона выражается кинетическим уравнением второго порядка в соответствии с уравнением лимитирующей стадии:
Химическая кинетика. Лаб. практикум |
67 |

2.ЛАБОРАТОРНЫЕ РАБОТЫ
2.6.Основные положения катализа
dC |
= k (C0 – Cх)(C0H+ + Cx), |
(23) |
d |
|
|
где C0 – начальная концентрация ацетона; C0H+ – начальная концентрация ионов водорода (кислоты); Cх – концентрация ацетона, подвергающаяся пре-
вращению за время (убыль концентрации); – время от начала реакции до данного измерения.
Интегрирование выражения (23) дает уравнение константы скорости процесса:
k |
2,303 |
lg |
C0 C0H |
Cx |
|
|
|
|
|
. |
(24) |
||
C0 C0H+ |
C0H C0 |
Cx |
||||
Для графической проверки это уравнение используют в линейной форме:
|
C0H Cx |
k C0 C0H ln |
C |
|
|
|
|
|
|
ln |
|
0 |
. |
|
|
|
(25) |
||
C0 Cx |
|
|
|
|
|||||
|
|
C0H |
|
|
|
|
|||
Дляэтогостроятграфикизависимостивкоординатах ln |
C0H Cx |
f ( ) . |
|||||||
|
C |
C |
x |
|
|||||
|
|
|
|
0 |
|
|
|
||
Экспериментальнаячасть
Реактивы, приборы, посуда: раствор соляной кислоты, раствор КI 4 %-й, ацетон, уксусная кислота 3 N, ацетат натрия (CH3COONa), колбы объемом 50 или 100 мл, спектрофотометр «SPEKOL-1300».
Последовательность выполнения работы. В работе исследуется за-
висимость константы скорости реакции от начальной концентрации катализатора (по указанию преподавателя), а также изучается первичный и вторичный солевой эффект.
Эксперимент проводят при четырех разных концентрациях соляной кислоты. В каждую из четырех мерных колб объемом по 100 мл вливают 12,5 мл 0,1 N раствора йода в 4 %-м растворе КI, прибавляют к нему 12,5 мл соляной кислоты с концентрацией от 0,1 до 0,9 N (по указанию преподавателя) и разбавляют дистиллированной водой, не доводя до метки. Затем вливают 1 мл ацетона и начинают отсчет времени. Момент вливания ацетона принимают за начало реакции. Доводят полученный раствор до метки водой. Аналогичную процедуру проделывают с остальными растворами йода с интервалом в 2-3 мин.
Первую пробу объемом 2 мл берут сразу же после доведения смеси до метки взбалтывания. Последующие пробы того же объема целесообразно
Химическая кинетика. Лаб. практикум |
68 |

2.ЛАБОРАТОРНЫЕ РАБОТЫ
2.6.Основные положения катализа
брать через каждые 15–20 мин (всего 8–10 проб). Чем меньше концентрация кислоты, тем реже следует брать пробы.
Анализ проб проводят с помощью спектрофотометра «SPEKOL-1300». Для этого изначально строят градуировочный график. Исследуемую пробу заливают в кювету, помещают в кюветное отделение прибора и измеряют оптическую плотность раствора (см. прил. 3, п. 5). Определяемую концентрацию рассчитывают по формуле
CONC ABC B K ,
где CONC – концентрация определяемого вещества в пробе; ABS – оптическая плотность пробы; В – угловой коэффициент уравнения градуировки; K – свободный член уравнения градуировки.
Подробная инструкция по работе с прибором «SPEKOL-1300» дана в прил. 3.
1.Расчет константы скорости проводят для каждой пробы и находят среднее значение.
2.Используя линейную форму уравнения (25), строят графики в коор-
динатах lg |
C |
Cx |
. По наклону каждой прямой находят значения кон- |
||
0H |
|
|
|||
C |
C |
x |
|||
|
|
||||
|
0 |
|
|
||
стант скорости и сравнивают с рассчитанными kср. Исходное количество аце-
тона C0 узнают по навеске, ионов водорода C0H+ – по количеству взятого раствора кислоты определенной концентрации, количество израсходованного ацетона Cx – по убыли йода, установленной по оптической плотности раствора.
3. Строят график зависимости константы скорости от концентрации кислоты.
4. Данные эксперимента и расчеты помещают в табл. 13.
Таблица 13
Длина волны, нм … Толщина кюветы, мм … Нормальность кислоты – … N
№ |
τ |
nτ |
Cx |
lg |
C |
0 H |
Cx |
|
k |
kср |
ln kcp |
|
|
|
|
||||||||||
C0 Cx |
||||||||||||
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
Изучение солевых эффектов проходит в два этапа: сначала исследуют первичный солевой эффект, затем – вторичный.
Первичный солевой эффект заключается в увеличении каталитического действия кислоты при прибавлении нейтральной соли, не имеющей общего аниона с катализирующей реакцию кислотой.
При постоянной начальной концентрации соляной кислоты проводят 5 опытов. Первый из них – без добавок соли, а последующие – с добавкой
Химическая кинетика. Лаб. практикум |
69 |

2.ЛАБОРАТОРНЫЕ РАБОТЫ
2.6.Основные положения катализа
соли в таком количестве, чтобы концентрация ее в реакционной смеси изменялась в пределах от 0,025 до 0,1 N. Полученные опытные и расчетные данные помещают в табл. 13.
Строят зависимости:
1)nτ = f(τ) (для нахождения n0);
2)константы скорости реакции от концентрации соли.
Вторичный солевой эффект заключается в том, что при добавлении в реакционную смесь соли катализирующей кислоты каталитическое действие последней может сначала уменьшаться, а затем возрастать с увеличением концентрации соли.
Катализатором здесь служит 3,0 N уксусная кислота (для всех опытов концентрация кислоты остается постоянной), добавками – уксусно-кислый натрий. При постоянной температуре проводят 8–10 опытов, причем максимальная концентрация соли в реагирующей смеси не должна превышать 0,2 N. Несколько опытов должны быть сделаны в пределах до 0,02 N концентрации соли, т.к. характерный для изучаемого эффекта минимум наблюдается примерно при 0,01 N соли. Полученные данные заносят в табл. 13, а также представляют в виде графиков.
Для подготовки к защите лабораторной работы можно воспользоваться следующей литературой: [1, с. 143–150, 183–186; 4, с. 208–217; 17, с. 145–147, 238–241].
Контрольныевопросыизадания
1.Почему скорость реакции йодирования ацетона не зависит от концентрации йода?
2.Получите соотношение (24) интегрированием уравнения (23).
3.Какие координаты, кроме предложенных в уравнении (25), можно использовать для графической проверки уравнения (24)?
4. Как влияет исходная концентрация реагентов на ход графиков lg C0H Cx , ?
5.Какие соли необходимо добавлять в реакционную смесь для изучения солевых эффектов?
6.Какой вид имеет кинетическая кривая автокаталитической реакции?
7.В чем заключается причина первичного солевого эффекта?
8.Выведите уравнение, описывающее зависимость константы скорости реакции от ионной силы раствора, используя уравнение Бренстеда – Бьеррума.
Химическая кинетика. Лаб. практикум |
70 |
2.ЛАБОРАТОРНЫЕ РАБОТЫ
2.6.Основные положения катализа
Лабораторная работа 11
Изучениекинетикикаталитическихреакций напримереразложенияперекисиводорода
Цельизадачиработы
Цель работы – изучение кинетики гомогенно-каталитического и гете- рогенно-каталитического разложения перекиси водорода. В задачи работы входят ознакомление с газометрическим методом изучения кинетики реакций, получение зависимости константы скорости от концентрации катализатора и определение константы равновесия реакции в гомогенно-каталитическом процессе, а также нахождение порядка реакции в процессе гетерогеннокаталитического распада перекиси водорода на активированном угле.
Теоретическоевведение
Перекись водорода в водных растворах самопроизвольно медленно разлагается по уравнению
Н2О2 Н2О + 0,5О2
Реакция заметно протекает при комнатной температуре только в присутствии катализаторов. Ими могут быть некоторые катионы и анионы в растворе, а также ряд твердых веществ. В соответствии с этим катализ разложения Н2О2 подразделяется на гомогенный и гетерогенный.
Гомогенно-каталитический распад Н2О2 в присутствии ионов Сr2О72-.
Разложение перекиси водорода в растворе под действием ионов Сr2О72- происходит в две стадии. В первой стадии реакции обратимо образуется промежуточное соединение:
2Н2О2 + Сr2О72- Cr2О92- + 2Н2О
Далее оно необратимо распадается с выделением кислорода и исходного ио-
на Сr2О72-.
Полагая, что лимитирующей стадией процесса является относительно медленный распад промежуточного соединения, общую скорость процесса можно считать пропорциональной концентрации промежуточного вещества:
|
d H2O2 |
k |
Cr O2 |
, |
|
||||
|
d |
|
расп 2 9 |
|
|
|
|
|
где kрасп – константа скорости распада промежуточного вещества ( Cr2 O92 ).
Концентрацию промежуточного вещества можно найти, используя константу равновесия первой стадии:
Химическая кинетика. Лаб. практикум |
71 |

2.ЛАБОРАТОРНЫЕ РАБОТЫ
2.6.Основные положения катализа
|
|
2- |
|
|
|
|
|
|
Kр |
Cr2O9 |
|
|
|
, |
(26) |
||
|
2- |
|
2- |
|
||||
|
|
|
||||||
|
H2O2 Cr2O7 |
Cr2O9 |
|
|
||||
где [Cr2O72-] – исходная концентрация катализатора; ([Cr2O72-] – [Cr2O92-]) – концентрация катализатора при равновесии. Вода находится в большом избытке, и ее концентрацию можно считать постоянной.
Из уравнения (26) получаем
|
|
2 |
|
Kр H2O2 |
2 |
|
2 |
|
|
|
|
|
|||
|
|
|
Cr2O7 |
|
|
|
|
|
|||||||
|
Cr2O9 |
|
|
|
|
|
|
|
|
, |
|
|
|
||
|
|
|
|
|
|
2 |
|
|
|
|
|||||
|
|
|
|
|
|
1 Kр H2O2 |
|
|
|
|
|
||||
откуда |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
d H2O2 |
|
|
|
Kр H2O2 |
2 |
|
2- |
|
|
|||||
|
kрасп |
|
Cr2O7 |
|
. |
(27) |
|||||||||
|
d |
1 Kр |
H2O2 2 |
|
|
|
|||||||||
|
|
|
|
|
|
|
|
|
|
||||||
Из последнего уравнения следует, во-первых, что скорость процесса пропорциональна начальной концентрации катализатора и, во-вторых, что в общем случае порядок реакции дробный и может изменяться от 0-го до 2-го.
Действительно, если равновесие сдвинуть в сторону образования промежуточного продукта, т.е. Kp[H2O2] >> 1, то порядок реакции равен нулевому по исходному веществу:
|
d H2O2 |
k |
расп |
Cr O2 . |
|
||||
|
d |
|
2 7 |
|
|
|
|
|
Когда Kp[H2O2] << 1, т.е. равновесие сдвинуто в сторону исходного вещества,
|
d H2O2 |
2 |
|
2 |
|
|
kрасп Kр H2O2 |
||
d |
Cr2O7 |
|||
|
|
|
|
|
и порядок реакции будет равен 2.
Поскольку сдвиг равновесия в ту или иную сторону зависит от температуры, то и порядок реакции изменяется с температурой. Уравнение (27) можно преобразовать в линейную форму, взяв обратное значение скорости:
|
|
1 |
|
|
|
|
1 |
|
|
1 |
|
1 |
|
|
. |
|
d H |
O |
|
|
|
2 |
2 |
|
2 |
||||||
|
|
|
|
|
H2O2 |
|
|
||||||||
|
|
2 |
2 |
|
|
|
kрасп Kр Cr2O7 |
|
|
kрасп Cr2O7 |
|
|
|||
|
|
d |
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
Химическая кинетика. Лаб. практикум |
72 |

2.ЛАБОРАТОРНЫЕ РАБОТЫ
2.6.Основные положения катализа
|
|
1 |
|
|
|
|
1 |
|
|
|
Построив график в координатах |
|
|
|
|
|
f |
|
|
|
, по тан- |
d H2O2 |
|
|
H2O2 |
2 |
||||||
|
|
dt |
|
|
|
|||||
|
|
|
|
|
|
|
|
|
||
генсу угла наклона прямой можно найти произведение kрасп Kр , |
а по отрезку |
|||||||||
на оси ординат – значение kрасп .
Таким образом, из кинетического опыта можно определить константу равновесия и константу скорости реакции.
Гетерогенно-каталитический распад Н2О2 на активированном угле.
Реакция разложения Н2О2 на активированном угле идет большей частью по первому или близкому к первому (0,7–1,2) порядку. Поэтому для оценки скорости реакции можно пользоваться кинетическим уравнением первого порядка:
d H2O2 k H2O2 . d
Так как изменение концентрации Н2О2 прямо пропорционально количеству выделяющегося кислорода, то при газометрическом измерении скорости реакции константу скорости удобнее определить через объем выделившегося О2:
k |
2,3 |
lg |
V |
, |
|
|
V |
V |
|||
|
|
|
|
|
|
где V – общий объем выделившегося кислорода; V – объем О2, выделившийся к моменту времени .
Экспериментальнаячасть
Реактивы, приборы, посуда: раствор перекиси водорода (Н2О2) 0,1–0,2 N, раствор перманганата калия (KMnO4) 0,1 N, серная кислота (Н2SO4) 20 %-я, раствор бихромата калия (К2Сr2О7) 0,05–0,2 N, активированный уголь, круглодонная колба с плотно закрывающейся резиновой пробкой с капилляром, установка для изучения каталитического разложения.
Последовательность выполнения работы. Одним из наиболее рас-
пространенных методов изучения кинетики разложения перекиси водорода является газометрический метод, который используется в нашей работе в обоих случаях.
Установка представлена на рис. 10 (цифрой 1 обозначен реакционный сосуд).
Химическая кинетика. Лаб. практикум |
73 |

2.ЛАБОРАТОРНЫЕ РАБОТЫ
2.6.Основные положения катализа
Рис. 10. Установка для изучения кинетики каталитических реакций
При снятии отсчетов по бюретке 2 необходимо приводить к одному уровню мениски жидкости в бюретке и уравнительной склянке 3, перемещая последнюю. Перед опытом необходимо проверить установку на герметичность. Для этого опускают уравнительную склянку ниже уровня бюретки. Уровень воды в бюретке при этом опустится и установится на некотором делении. Если это положение не изменится в течение 3 мин, установку можно считать герметичной. В противном случае уровень воды в бюретке будет непрерывно расти.
В работе следует изучить реакцию распада перекиси водорода на жидком и твердом катализаторах.
1. Изучение гомогенно-каталитического разложения перекиси водорода.
Цель данной работы – получение зависимости константы скорости от концентрации катализатора и нахождение константы равновесия реакции.
Нужно приготовить раствор перекиси водорода с концентрацией в пределах 0,1–0,2 (150–200 мл). Точная концентрация Н2О2 определяется титрованием перманганатом калия. Для этого наливают пипеткой 2–3 мл рабочего раствора ( 1 N) и 1–2 мл 20 %-го раствора Н2SO4. Титруют 0,1 N раствором КМnO4 до появления слабо-розового окрашивания.
При этом протекает реакция
2MnO4- + 6H+ + 5H2O2 2Mn2+ + 8H2O + 5O2
Знание точной концентрации раствора Н2О2 необходимо для сравнения опытного конечного объема выделившегося кислорода и теоретического, которое можно рассчитать по уравнению Клапейрона-Менделеева с учетом давления насыщенного пара воды при температуре опыта. В каждом опыте используют одно и то же количество раствора перекиси водорода в пределах 20–25 мл. Проводят 2-3 опыта, варьируя концентрацию раствора катализатора К2Сr2О7 в пределах 0,05–0,2 N, сохраняя одинаковый добавляемый объем 3 мл. Опытные данные вносят в табл. 14 и представляют графически:
Химическая кинетика. Лаб. практикум |
74 |

2.ЛАБОРАТОРНЫЕ РАБОТЫ
2.6.Основные положения катализа
1)кинетические кривые VO2 = f( ) и CH2O2 = f( ) для всех концентраций
катализатора. По этим кривым графическим дифференцированием определяют скорость реакции;
|
|
1 |
|
|
|
1 |
|
|
2) график зависимости |
|
|
|
|
f |
|
|
для определения |
d H2O2 |
|
|
H2O2 2 |
|||||
|
|
dt |
|
|
|
|||
kрасп и Kр ; |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3) график зависимости kрасп f Cr2O72 .
2. Изучениегетерогенно-каталитическогоразложенияперекисиводорода.
Проводится три опыта с разными навесками активированного угля. Точную концентрацию 1 N раствора Н2О2 определяют титрованием,
как описано в п. 1. Затем в колбу заливают 20 мл раствора перекиси водорода, засыпают навеску активированного угля (0,25–1,0 г – задается преподавателем), быстро закрывают пробкой на отводящем шланге, одновременно включают секундомер, начинают интенсивно встряхивать реакционный сосуд (это производят в течение всего опыта, чтобы исключить влияние процесса диффузии на кинетику реакции) и приступают к отсчетам объема выделившегося кислорода по шкале бюретки.
Опытные данные заносят в табл. 15 и представляют графически:
1)кинетические кривые VO2 f ( ) и VH2O2 f ( ) ;
2)графики для определения порядков реакции в координатах lgv f (lgC) по графам 1 и 4 табл. 15
.
Химическая кинетика. Лаб. практикум |
75 |

2.ЛАБОРАТОРНЫЕ РАБОТЫ
2.6.Основные положения катализа
Таблица 14
С = …, СН2О2 = … N, VH2O2 = … мл, VK2Cr2O7 = … мл, концентрация исходного раствора катализатора К2Cr2O7
Концентра- |
|
|
Количество, моль |
|
|
Концентрация |
|
|
|
|
|
|
1 |
|
|
|
|
|
|
|
||||||||
ция раство- |
, |
Объем |
выде- |
оставшейся |
оставшейся перекиси |
|
|
Скорость раз- |
|
|
|
1 |
|
|
|
К, |
|
|||||||||||
ра катализа- |
кислорода |
ленного |
перекиси |
|
|
ложения Н2О2 |
|
|
|
k, с-1 |
2 |
|||||||||||||||||
|
|
мин |
|
|
nH2O2 = |
CH |
|
|
|
nH2O2 100 |
|
|
|
= СН |
|
|
|
H2O2 |
|
2 |
|
|
|
(моль/л) |
|
|||
тора в реак- |
|
V, мл |
кисло- |
O |
|
VK Cr O |
|
VH |
|
|
|
О |
/ |
|
|
|
С Н |
О |
|
|
|
|
||||||
|
|
|
O |
|
|
|
|
|
|
|
||||||||||||||||||
торе Cr2O7 |
2- |
|
|
рода nO2 |
= 2[nO2 – nO2 ] |
2 |
|
2 |
|
7 |
|
2 |
2 |
|
|
|
2 |
|
2 |
|
|
|
||||||
|
|
|
|
|
|
|
2 |
2 |
2 |
|
2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Таблица 15 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
СН2О2 = … N, VН2О2 = … мл, количество угля = … г |
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
, |
Объем ки- |
Количество, моль |
Концентрация |
Скорость раз- |
|
|
|
lg |
|
|
Порядок |
||
выделившегося |
оставшейся пе- |
оставшейся |
ложения Н2О2 |
lg CH |
|
|
|
k, c-1 |
|||||
слорода |
O |
|
|
||||||||||
мин |
V, мл |
кислорода nO2 |
рекиси nH2O2 |
перекиси СН2О2 |
|
2 |
|
2 |
|
|
реакции n |
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Химическая кинетика. Лаб. практикум |
76 |

2.ЛАБОРАТОРНЫЕ РАБОТЫ
2.6.Основные положения катализа
Для получения воспроизводимых результатов рекомендуется все опыты дублировать два–три раза.
Для подготовки к защите лабораторной работы можно воспользоваться следующей литературой: [1, с. 124–143, 194–224; 4, с. 345–414; 13, с. 467–474; 19, с. 431–441, 454–472].
Контрольныевопросыизадания
1.Почему катализатор ускоряет реакцию?
2.Что означают термины «селективность» и «специфичность» катали-
затора?
3.Каковы механизмы гомогенно- и гетерогенно-каталитических про-
цессов?
4.Каков режим протекания гетерогенно-каталитического разложения Н2О2? Есть ли возможность изменить его?
5.Изобразите кривые изменения потенциальной энергии в каталитическом процессе.
6.Почему катализатор не смещает равновесие реакции?
7.Какие факторы определяют активность гетерогенного катализатора?
8.Покажите, как графически определить константу скорости и константу равновесия реакции гомогенно-каталитического разложения перекиси водорода. Объясните, почему график строится именно в таких координатах.
9.Из каких стадий состоит гетерогенно-каталитическая реакция? Каким образом определяют общую скорость такого процесса?
10.Перечислите возможные отличия гетерогенно-каталитических реакций, протекающих в жидкой фазе, от реакций в газовой фазе.
Химическая кинетика. Лаб. практикум |
77 |