

Genomics: The Science and Technology Behind the Human Genome Project. |
Charles R. Cantor, Cassandra L. Smith |
|
Copyright © 1999 John Wiley & Sons, Inc. |
|
ISBNs: 0-471-59908-5 (Hardback); 0-471-22056-6 (Electronic) |
12 Future DNA Sequencing without Length Fractionation
WHY |
TRY |
TO |
AVOID LENGTH FRACTIONATIONS? |
All |
but |
one |
of the methods we have described for DNA sequencing in Chapter 10 in- |
volved a gel electrophoretic fractionation of DNA. The exception used mass spectroscopy |
|||
instead of electrophoresis, but a length fractionation was still needed because all of the in-
formation about base location had been translated into fragment sizes prior |
to |
analysis. |
There are several motivations to try to get away from this basic paradigm |
and |
develop |
DNA sequencing methods that do not depend on size fractionation. First, size fractionation, except by mass spectroscopy, is really quite a slow and indirect method of reading sequence data. Second, fractionations are intrinsically hard to parallelize. Third, it is not obvious how fractionation methods could ever be used to look efficiently just at sequence differences, and most of the long-term future of DNA sequencing applications probably
lies in this key area of differential sequencing. This is true not only in the potential use of DNA sequencing for human diagnostics, but also for evolutionary applications, for popu-
lation genetic applications, and for ecological screening.
For all of these reasons there is considerable current interest in trying to develop entirely new approaches to DNA sequencing. Many of the techniques that will be men-
tioned in this chapter are going to be discussed only briefly. While they are probably capable of maturing into methods that can read DNA sequences, they are unlikely to do this soon enough, or ultimately efficiently enough, to be of much use for large-scale DNA sequence processing. However at least one of the second-generation methods treated in this chapter, sequencing by hybridization (SBH), does appear to offer a significant chance of making an impact on the current human genome project, and an even better chance of making a major impact on future DNA sequencing in clinical diagnostics.
SINGLE-MOLECULE |
SEQUENCING |
|
A number of different potential DNA sequencing methods require that data |
be obtained |
|
from one single molecule at a time. They include handling DNAs or their reaction prod- |
||
ucts in flow systems, or observing DNAs by microscopy. One approach that has been in- |
||
vestigated by Richard Keller, would use an exonuclease to degrade a tethered DNA con- |
||
tinually, and detect individual nucleotides as they are cleaved by the enzyme and liberated |
||
into a flowing stream. This method exploits the power of flow cytometry for very sensi- |
||
tive detection of |
a fluorescent target whose location is known |
rather precisely. A |
schematic illustration of this approach to single-molecule sequencing is given in Figure 12.1.
394

SINGLE-MOLECULE SEQUENCING |
395 |
U |
A |
|
Fluorescently |
||
G |
||
labeled |
A |
|
DNA |
C |
|
strand |
||
C |
||
U |
||
|
||
|
Exonuclease |
|
U |
|
Fluid
flow
 G
G
Cleaved, labeled nucleotides
|
G |
|
Focused |
|
C |
laser beam |
|
|
|
|
|
Spectral filter
 Photodetector
Photodetector
Computer
Figure 12.1 Schematic illustration of one approach to sequencing single DNA molecules in solution. Provided by Richard Keller.
To see all four bases in a single molecule, each would have to be labeled with a different fluorophore. There have been suggestions to use the intrinsic fluorescence of each base directly, but this fluorescence is weak, so it would require very sophisticated and expensive detection methods to be practical at the single-molecule limit. The challenge is to make fluorescent base analogs that are acceptable substrates for DNA polymerase. Substitution at every single nucleotide position must be accomplished. This requirement has been met with one and two bases, which is an impressive accomplishment. It remains
to be seen if it can be met with all four bases. The ideal exonuclease would liberate bases
in a kinetically smooth and rapid rate. It would be processive so that a single enzyme would suffice to degrade the DNA molecule. Otherwise, with the arrangement shown in
Figure 12.1, there would be pauses during which no product would be appearing, and one

396 |
FUTURE DNA SEQUENCING WITHOUT LENGTH FRACTIONATION |
|
|
|||
would constantly have to replenish the supply of enzyme as molecules fall off and are lost |
|
|||||
to |
the flowing stream. The properties |
of actual exonucleases, for the most |
part, are |
not |
||
this ideal, but they are rapid, and some are reasonably processive. |
|
|
|
|||
|
It is possible in a flowing stream to detect single fluorophores of the sort used to label |
|||||
nucleic acid bases, which is like that which would be used with a tethered DNA. Some |
||||||
typical results are illustrated in Figure 12.2. The issue that remains |
unresolved |
is the |
||||
chances of missing a base (a false negative) and the chances of seeing a noise fluctuation, |
||||||
by |
scattering from a microscopic dust |
particle, or whatever, that imitates a |
base when |
|||
none is present (a false positive). |
It is interesting to examine what |
the |
consequences |
|||
would be if, instead of one molecule, many were tethered together in the flowing stream, |
|
|||||
and exonucleases were allowed to process them all. If the digestion could be kept in syn- |
||||||
chrony, the signal-to-noise problem |
in detection would be alleviated |
considerably. |
||||
However, there is no way to keep the reactions |
in synchrony. The best one |
can |
do is |
to |
||
find |
a way to start the exonucleases synchronously |
and use the most processive |
enzymes |
|
||
available. Even in this case, however, there is an inevitable dephasing of the reaction, as it proceeds, because of the basic stochastic (noisy) nature of chemical reactions. Given
Figure 12.2 |
Typical data obtained in pilot experiments to test |
the scheme shown in Figure 12.1. |
Top panel: A |
dilute concentration of a labeled nucleotide is allowed |
to flow past the detector. |
Bottom panel: A control with no labeled nucleotides. |
|
|
SEQUENCING BY HIGH-RESOLUTION MICROSCOPY |
397 |
Figure 12.3 |
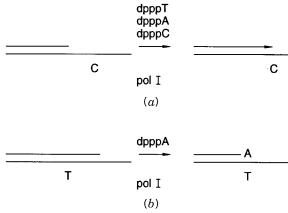
Plus-minus DNA sequencing. ( |
a ) Extension until a template coding for a missing |
dpppN is encountered. ( |
b ) Degradation of a chain until a specific nucleotide is reached. |
|
some microscopic rate constant for a reaction, at the level of individual molecules the ac- |
|
||||||||||||
tual reaction times vary in such a way that a distribution of rates actually contributes the |
|
||||||||||||
average value |
seen macroscopically. The reaction zone broadens like a random walk as |
|
|||||||||||
the |
reaction |
proceeds down the chain. Its width soon becomes more than several bases, |
|
|
|||||||||
even under ideal kinetic circumstances. |
|
|
|
|
|
|
|
|
|||||
|
One |
way to circumvent |
the |
statistical problems |
with |
sequencing |
by |
exonucleases |
|
||||
would be to find a method to stop the reaction at fixed points and then allow it to restart. |
|
||||||||||||
This, in essence, is what is |
done with DNA polymerase in |
plus-minus sequencing, the |
|
|
|||||||||
very earliest method used by Ray Wu. If one dpppN is left |
out, the reaction proceeds |
up |
|
||||||||||
to the place |
where this base is demanded by the template, and it stalls there |
(Fig. 12.3 |
a ). |
||||||||||
Adding the missing dpppN then |
allows the reaction to continue. If a DNA |
polymerase |
|
|
|||||||||
with |
a 3 |
-editing exonuclease activity is used, a similar result can be achieved by having |
|
||||||||||
only one dpppN present. In this case the enzyme degrades the 3 |
|
|
|
|
|
-end of a DNA chain, un- |
|||||||
til it reaches a place where the |
dpppN present is called for |
by |
the template |
(Fig. 12.3 |
b ). |
||||||||
As long |
as a |
sufficient supply of dpppN remains, the enzyme will stall at this position. |
|
||||||||||
These are useful tricks; they work well for sequencing, and there is no reason why they |
|
||||||||||||
could not be incorporated into strategies for sequencing a single molecule or small num- |
|
|
|||||||||||
bers |
of molecules. However, the major potential advantage of the original scheme pro- |
|
|
||||||||||
posed by Keller is speed, and steps that require changing substrates numerous times are |
|
||||||||||||
likely to slow down the whole process considerably. |
|
|
|
|
|
|
|
|
|||||
SEQUENCING BY HIGH-RESOLUTION MICROSCOPY |
|
|
|
|
|
|
|
|
|||||
One |
of |
the |
earliest attempts at the development of alternate DNA sequencing methods |
|
|
||||||||
was |
Michael |
Beer’s strategy for determining nucleic acid sequence by |
electron |
mi- |
|
||||||||
croscopy. Beer’s plan was to label individual bases with particular electron-dense heavy |
|
||||||||||||
metal clusters and then image these. Two problems made this approach unworkable. First, |
|
|
|||||||||||
the nucleic acids were labeled |
by |
covalent modification |
after |
DNA |
synthesis. This |
leads |
|
|
|||||
to less than |
perfect stoichiometry |
of the desired product, and it |
undoubtedly |
also |
leads |
to |
|
||||||

398 FUTURE DNA SEQUENCING WITHOUT LENGTH FRACTIONATION
some unwanted side reactions with other bases. The second problem is that sample dam-
age in the conventional electron microscope is considerable; this makes it very difficult to achieve accurate enough images to read the DNA sequence as the spacing between metal-
tagged bases, since the structure moves around in response to the electron beam of the molecule. This problem of molecular perturbation by microscopes remains with us today
as the greatest obstacle to using high-resolution microscopy for DNA sequencing. |
|
||||||
Currently |
a |
new |
generation |
of |
ultramicroscopes has reopened |
the issue of |
whether |
DNA could be sequenced, directly or indirectly, by looking at it. The new instruments are |
|||||||
scanning tip microscopes; the best studied of these are the scanning tunneling microscope |
|||||||
(STM) and the atomic force microscope (AFM). Both of these instruments read the sur- |
|||||||
faces of samples in much the same way that a blind person reads braille. The surface is |
|||||||
scanned with a |
sharp |
tip in a |
raster pattern, as shown schematically in Figure 12.4. In |
||||
AFM what leads to the image is the force between the tip and the sample. Van der Waals |
|||||||
forces will attract the tip to the surface at long distances and repel the tip |
at short dis- |
||||||
tances. What is |
usually done is to |
have a feedback loop via a |
piezoelectric |
device. This |
|||
can be used to place a force on the tip to keep its vertical position constant, and the voltage needed to accomplish this is measured. Alternatively, one can apply a constant force,
and measure the vertical displacement of the tip, for example, by bouncing a laser beam
off the tip and seeing where it is reflected to. In STM an electrical potential is maintained between the tip and the surface. This leads to a current flow from the tip to the surface at short distances. The current is dependent on the distance between the tip and the surface,
and the electrical properties of the surface. In practice, one can adjust the position of the tip to maintain a constant current, and measure the tip position, or keep the vertical height of the tip constant and measure the current.
For AFM or STM to be successful, very flat surfaces are required. With hard samples on such surfaces, atomic resolution is routinely observed, and even subatomic resolution
has been reported, where information about the distribution of electron density within the sample is uncovered. DNA is not a hard sample, and it does not easily adhere to most very flat surfaces. These difficulties have produced many frustrations in early attempts to image DNA by AFM or STM. In retrospect, most or all of the spectacular early pictures of DNA have been artifacts, perhaps caused by helixlike imperfections in the underlying surfaces. The best that can be said is that images that looked like DNA were rare and far between, and not generally reproducible. One problem that soon became quite apparent is
that the forces used in these early attempts were sufficient in most, if not all, cases to knock the DNA molecules off the surface being imaged.
Figure 12.4 Operating principle of a typical scanning tip microscope. In STM the electrical current between the tip and the surface is measured. In AFM the repulse force between the tip and the
surface is measured.

|
|
|
SEQUENCING BY HIGH-RESOLUTION MICROSCOPY |
399 |
||
More recent attempts to image DNA with scanning tip microscopes, particular with |
|
|
||||
AFM, have been more successful, at least in the sense that dense arrays of molecules can |
|
|
||||
be seen reproducibly. This is accomplished by using surfaces to which DNA adheres bet- |
|
|
||||
ter, like freshly cleaved mica, instead of the graphite or silicon surfaces |
used |
earlier. |
|
|||
Sharper tips give high enough resolution to be able to measure the lengths of the mole- |
|
|||||
cules reliably. The current images are, however, a long way from the resolution needed to |
|
|
||||
read the sequence directly by looking at the bases. A number of severe obstacles will have |
|
|
||||
to be overcome if this is ever to be done. First, the current images are mostly of double- |
|
|||||
stranded |
DNA. This is understandable since |
it is a much more regular structure, |
much |
|
|
|
more amenable to detection and quantitation in the microscope. However, in the double |
|
|
||||
strand, only short bits of sequence are readable from the outside, as one is forced to do in |
|
|||||
AFM. This will lead to a difficult, but apparently not insurmountable, sequence recon- |
|
|||||
struction problem where data from many molecules will have to be combined to synthe- |
|
|
||||
size the final sequence. A second problem is that the DNA molecules could still be dis- |
|
|
||||
torted quite a bit as the tip of the microscope moves over them. This may or may not be |
|
|
||||
alleviated by newer microscope designs that would allow lower forces to be used. |
|
|
|
|||
A third problem with AFMs is |
that the image seen is a convolution of the shape of the |
|
|
|||
tip and the shape of the molecule, as shown in Figure 12.5. Thus, unless very sharp |
tips |
|
||||
can be made, or tips of known shape, it can be difficult with a soft, deformable molecule |
|
|||||
to deconvolute the image and see the true shape of the DNA. One approach to circumvent |
|
|
|
|||
many of these difficulties would be to label the DNA with base-specific reagents that are |
|
|
||||
more distinctive either in AFM, where larger, specific shapes could be used, or in STM, |
|
|||||
where labels with different electrical properties might serve. As a test of this, and to make |
|
|||||
sure DNA |
imaging was now reliable |
in the AFM, proteins were attached to the ends |
of |
|
|
|
DNAs before AFM imaging. Two examples of |
the sorts of images seen are shown in |
|
|
|||
Figure 12.6. The protein used, purely because it was available and of a size that made it |
|
|||||
easy to distinguish from DNA, was a chimera between streptavidin and a fragment of |
|
|
||||
staphylococcal protein A, which was already introduced in Chapter 4, where it was used |
|
|
||||
for immunoPCR. The DNA was biotinylated, either on one or both ends. Two different |
|
|
||||
lengths |
of DNAs were used, and |
since streptavidin is tetrameric, the resulting |
images |
|
||
show a progression of structures from DNA monomers up to trimers. Because of the na- |
|
|
|
|||
ture of these structures, the proper measured lengths of the DNAs within them, and the |
|
|
||||
expected |
height difference between |
the protein label at the ends or vertices of the |
DNA |
|
|
|
and the DNA itself, one can be very confident that these are true images of DNA and protein. However, the resolution is still far too low to allow sequencing.
Figure 12.5 In scanning tip microscopy, what is actually measured is a convolution of the shape of the object and the shape of the tip.

400 FUTURE DNA SEQUENCING WITHOUT LENGTH FRACTIONATION
Figure 12.6 Two typical AFM images of short-end biotinylated DNA molecules labeled at one or both ends by a chimeric protein fusion of streptavidin and staphylococcal protein A. Since the streptavidin is tetrameric, one can see figures representing more than one DNA molecule bound to
the same streptavidin. Reproduced from Murray et al. (1993).
It should be possible to use progressively smaller labels and to increase the resulting resolution. Whether this will lead to direct AFM DNA sequencing soon is anyone’s guess.
If it does, a real advantage is that one could sequence a wide variety of different molecules in a single experiment without the need to clone or fractionate. The labeling would almost certainly be introduced by PCR using analogs of the four bases. This will be much more accurate than the original chemical modification methods used for electron microscopy. However, the resulting images, as elegant as they may look some day, might have to be analyzed as images to extract the DNA sequence data. By current methods this
could become a serious bottleneck. What is still needed is a way to direct the tip of the microscope so that it tracks just over the DNA molecule of interest, rather than scanning a grid that is mostly background. If this can somehow be achieved, the problem of image analysis ought to become much simpler, and the rate of image acquisition also ought to
be increased considerably.
STEPWISE ENZYMATIC SEQUENCING
A major success story in the history of protein sequencing was the development of stepwise chemical degradation. Amino acid residues are removed one at a time from one end
of the polypeptide chain and their identity is determined successively. Automated

|
|
|
|
|
STEPWISE ENZYMATIC SEQUENCING |
401 |
||||
Edmond degradation currently provides our main source of direct protein sequence data. |
|
|
||||||||
The yield in each step is the critical variable, since it determines how far from the original |
|
|||||||||
end the sequence can be read. Comparable chemical approaches for DNA or RNA se- |
|
|
|
|||||||
quencing have not been terribly successful. Recently, however, several |
stepwise |
enzy- |
|
|||||||
matic sequencing approaches |
have been suggested. As individual processes, they do |
not |
|
|
||||||
at first glance seem all that attractive. However, they have the potential to be implemented |
|
|||||||||
in massively parallel configurations, which, if successful, could ultimately provide very |
|
|||||||||
high throughput. These schemes are distinct from the single molecule methods described |
|
|
||||||||
earlier in that any desired number of target molecules of each type can be employed. Thus |
|
|
||||||||
detection sensitivity is not an issue here. |
|
|
|
|
|
|
||||
One strategy, developed by Mathias Uhlen, is to divide the sample into four separate |
|
|||||||||
wells, |
each |
containing |
a |
DNA polymerase without a 3 |
|
|
|
|
-proofreading activity. To each |
|
well one of the four dpppN’s is added. Chain extension will occur only in |
one well with |
|
||||||||
the concomitant release of pyrophosphate (Fig. 12.7). This product can be detected with |
|
|||||||||
great sensitivity; for example, it can be enzymatically converted to ATP, and that can be |
|
|||||||||
measured using luciferase, to generate a chemiluminescent signal. The amount of light |
|
|
||||||||
emitted |
is proportional |
to |
the amount of ATP made. Thus one can quantitate |
the amount |
|
|
||||
of dpppN incorporated and determine, within limits, how many units the chain was ex- |
|
|
||||||||
tended by. Sample from the well that was successfully extended is then divided into four |
|
|||||||||
new wells, and the process is repeated. Actually three wells would |
suffice, |
since |
one |
|
||||||
knows that the next base is not the same as the one or ones just added, but it is probably |
|
|||||||||
good to have the fourth as an internal control. One obvious complication with the scheme |
|
|
||||||||
is that the sample keeps getting divided, so one has to either start with |
a large |
amount of |
|
|||||||
it or have a |
sensitive enough assay that only a small aliquot can be removed |
and assayed. |
|
|
||||||
A variation on this basic approach adds dpppNs in a cyclical order. This avoids the prob- |
|
|
||||||||
lem of sample subdivision. It appears to have considerable promise. The method is called |
|
|
||||||||
pyrosequencing. |
|
|
|
|
|
|
|
|
||
A second strategy is similar in spirit but uses dideoxy pppN’s. This is shown in Figure |
|
|||||||||
12.8. In four separate wells containing target DNA and DNA polymerase is added one of |
|
|
||||||||
the ddpppN’s carrying a label. Alternatively, one could use a single well and a mixture of |
|
|||||||||
four different fluorescently labeled ddpppN’s. Only one of the ddpppN’s becomes incor- |
|
|||||||||
porated. From the location of the well, or the color, the identity of the base just added is |
|
|||||||||
known. The base just added is now removed by treating with the 3 |
|
|
|
|
-editing exonuclease |
|
||||
activity |
of |
DNA polymerase |
I in the presence of all of the dpppN’s |
except the |
one |
just |
|
|||
Figure 12.7 |
One scheme for stepwise enzymatic |
DNA |
sequencing. Here, when a particular base |
is added, pyrophosphatase is used to synthesize ATP from |
the |
pyrophosphate (pp) released, and the |
|
ATP in turn is used to generate a chemiluminescent signal by serving as a substrate for the enzyme, luciferase.

402 FUTURE DNA SEQUENCING WITHOUT LENGTH FRACTIONATION
Figure 12.8 A second scheme for stepwise enzymatic DNA sequencing. This is similar in spirit to plus-minus DNA sequencing illustrated in Figure 12.3. It uses fluorescent terminators, ddpppN*, like those employed in conventional automated Sanger sequencing.
added. Next the labeled ddpN |
just removed is replaced by an unlabeled dpppN by using |
|
DNA polymerase with only this particular dpppN present. Then the process is repeated. |
||
This |
scheme avoids the sample division or aliquoting problem of the previous strategy. |
|
|
To detect a run of the |
same base, one will have to be able to vary the scheme. What |
will |
happen in this case is |
that the exonuclease treatment will degrade the chain back to |
the location of the first base in the run. To determine the length of the run, one possible approach is to add a labeled analog of the particular dpppN involved in the run, in the presence of DNA polymerase, and detect the amount of synthesis by quantitating the incorporation of label. Then the entire block of labeled dpN’s has to be removed, replaced by unlabeled dpN’s, and next base after the block can now be determined. This is an unfortunate complication. However, in principle, the entire scheme could be set up in a mi-
crotitre plate format and run in |
a very parallel way. As in the first scheme |
the whole |
process would be best carried out |
in a solid state sequencing format so that the DNA |
|
could be purified away from small |
molecules and enzymes easily and efficiently |
after |
each step. |
|
|
A third strategy, has not been tested to our knowledge, because it depends on finding a |
||
dpppNx derivative that has two special properties. Like a ddpppN the derivative must not be extendable by DNA polymerase. However, there must be a way to change the dpNx af-
ter incorporation |
into the DNA chain so that now it is extendable. The scheme then, as |
shown in Figure |
12.9, is to add dpppNx to the target in four separate tubes. Whichever |
Figure 12.9 A third, as yet unrealized scheme for stepwise enzymatic DNA sequencing. Here the
key ingrediant would be a 3 |
-blocked pppNx |
that could be deblocked after each single base |
elongation step. Incorporation could be detected by pyrophosphate release as in Figure |
||
12.7 or by the use of a fluorescent blocking agent, x |
. |
|
|
|
|
|
|
DNA SEQUENCING BY HYBRIDIZATION (SBH) |
403 |
|||
one extends could be determined by a pyrophosphatase assay as in the first scheme. Then |
|
||||||||
the incorporated dpNx is converted to dpN, and the process continued. One candidate, in |
|
||||||||
principle, for the desired |
compound is dpppNp, which, after incorporation of the dpNp |
|
|||||||
into DNA could be converted to dpN by alkaline phosphatase. This latter step certainly |
|
||||||||
works; however, it is uncertain how well DNA polymerase will utilize compounds like |
|
||||||||
dpppNp’s. Apparently |
quite |
|
a few other reversible dpppN |
derivatives have been tried as |
|
||||
DNA polymerase substrates without much success thus far. |
This is a pity because the |
|
|||||||
scheme has real appeal. |
|
|
|
|
|
|
|
|
|
In deciding how, eventually, to implement any of the above schemes in an automated |
|
||||||||
fashion, one needs to consider an interesting trade-off between the time to sequence one |
|
||||||||
sample and the number of samples that can be handled simultaneously. Instead of trying |
|
||||||||
all four single base additions simultaneously, they could be tried one at a time, in a cycli- |
|
||||||||
cal pattern, say A, then |
G, then C, then T, as in |
pyrosequencing. With an immobilized |
|
||||||
DNA sample, the target is simply moved from one set of reagents, after washing, to an- |
|
||||||||
other, and the point where |
|
positive |
incorporation occurs |
is recorded. The advantage of |
|
||||
this is that the logistics |
and design of the system become much simpler, particularly for |
|
|||||||
the cases where pyrophosphate release is measured. It will take four times as long to |
|
||||||||
complete a given length of DNA sequence, but one could handle precisely four times as |
|
||||||||
many sequences simultaneously. |
|
|
|
|
|
|
|||
DNA SEQUENCING BY HYBRIDIZATION (SBH) |
|
|
|
|
|||||
We will devote the rest of this chapter to a number of approaches for determining the se- |
|
||||||||
quence of DNA by hybridization. In all of these approaches one uses relatively short |
|
||||||||
oligonucleotides as probes to determine whether the target contains the precise comple- |
|
||||||||
mentary sequences. Essentially SBH reads a word at a time rather than a letter at a time. |
|
||||||||
Intuitively this is quite |
efficient; it is after all the way written |
language is usually read. |
|
||||||
For reasons that will become readily apparent, attempts to perform SBH have usually fo- |
|
||||||||
cused on oligonucleotides with 6 to 10 bases. The conception of SBH appears to have had |
|
||||||||
at least four independent origins. Many groups are now working to develop an efficient |
|
||||||||
practical scheme to implement SBH. |
|
|
|
|
|
|
|||
There seems to be a consensus that SBH will eventually work well for some high- |
|
||||||||
throughput DNA sequencing applications, like sequence comparison, sequence checking, |
|
||||||||
and clinical diagnostic sequencing. In all of these cases one |
is not |
trying to determine |
|
||||||
large tracts of sequence de novo; instead, the targets of interest are mostly small differ- |
|
||||||||
ences between a known or expected sequence, and what has actually been found. There is |
|
||||||||
also agreement that SBH will work for determining partial sequences, for fingerprinting, |
|
||||||||
and for mapping. However, SBH may not work for direct complete de novo sequencing |
|
||||||||
unless some of the enhancements or variations that have been proposed to circumvent a |
|
||||||||
number of problems turn out to work in practice. |
|
|
|
|
|||||
The two critical features of SBH are illustrated in Figures 12.10 and 12.11. As we |
|
||||||||
demonstrated in Chapter |
3, |
|
the stability of a perfectly matched duplex is greater than an |
|
|||||
end mismatched duplex, and much greater than a duplex with an internal mismatch (Fig. |
|
||||||||
12.10). Thus a key step in SBH is finding conditions where there is excellent discrimina- |
|
||||||||
tion between perfect matches and mismatches. An immediate |
problem is that for a se- |
|
|||||||
quence of length |
n, |
there is only one perfect match, there are six possible end mismatches |
|
||||||
(each base on each end of |
the target can be |
any one |
of the |
three |
noncomplementary |
|
|||
bases), and there are |
3( |
|
n |
2) |
possible internal |
mismatches. Unless the discrimination is |
|
||