

AUTOMATED DNA SEQUENCING CHEMISTRY |
345 |
Figure 10.15 Example of background in DNA sequencing ladders generated by mispriming.
strand which carried the consensus sequence YGN1–2AR, where Y and R are complementary. Note that this motif is contained in the extraordinarily stable hairpin just described above. Now that the prevalence of this motif in compressions is understood, it can
be used to correct the misread sequence as shown by the example in Figure 10.14.
Other sources of error in DNA sequencing are caused by |
mispriming. Not uncom- |
||
monly, there will be a secondary site on the template where the primer can bind and be |
|||
extended by the polymerase (Fig. 10.15). This adds a bit of low-level, specific noise to the |
|||
primary sequencing data. Another source of ambiguity arises when the sample is het- |
|||
erozygous or a mixture. The basic point is that most of |
these |
errors can be partially or |
|
even totally corrected if the software is clever enough to search out these possibilities. As |
|||
more raw sequence data are obtained, and ultimately corrected into finished sequence, it |
|||
should be possible to go back to the raw data and refine the algorithms used to process it. |
|||
In short, the automated analysis of DNA sequencing data ought to be able to improve it- |
|||
self continually with time. The ideal software, which does not |
yet exist, |
would actually |
|
give use the probability of each of the four bases occurring at a given position. At a given |
|||
site the result might be |
|
|
|
|
A |
0.01 |
|
|
G 0.98 |
|
|
|
C |
0.00 |
|
|
T |
0.01 |
|
This would be the best data to feed back into artificial intelligence |
approaches to refine |
||
the software further. A nice step in this direction is Phil Green’s phred algorithm which |
|||
automatically calls sequences and assigns a quality score, |
|
|
q, to each base, |
q |
10 log |
p |
|
where p is the estimated error probability for that base. Hence phred scores of 30 or better indicate sequences that are likely to be perfect.
AUTOMATED DNA SEQUENCING CHEMISTRY
The one remaining area we need to describe, where great progress has been made is the automated preparation of DNA for sequencing. The most success appears to be seen with solid state DNA preparations. These were developed by Mathias Uhlen, and a recent in-

346 DNA SEQUENCING: CURRENT TACTICS
Figure 10.16 |
Two solid state DNA sequencing schemes. |
(a ) From a plasmid DNA miniprep. ( |
b ) |
From DNA prepared by PCR. Provided by Mathias Uhlen. See Holtman et al. (1989).

AUTOMATED DNA SEQUENCING CHEMISTRY |
347 |
teresting modification has been accomplished by Ulf Landegren. In both methods the idea is to capture DNA onto a solid surface via streptavidin-biotin technology, and then do at least one strand of the DNA sequencing on that surface. Two schemes developed by
Uhlen are illustrated in Figure 10.16. They are pretty much self-explanatory. The Uhlen implementation of these solid state preparations uses magnetic microbeads containing immobilized streptavidin. The DNA is biotinylated either by filling in a restriction site with a biotinylated base analog or by using a biotinylated PCR primer. Once the duplex DNA
is captured, the nontethered strand is removed by alkali. An essential aspect of the procedure is that the streptavidin-biotin link is resistant to the harsh alkali treatment needed to melt the DNA. Sequencing chemistry is then carried out on the immobilized DNA strand
that remains. If desired, the strand released into solution can also be subsequently captured in a different way and sequenced. The great advantage of this approach is the ease with which it can be automated and the very clean DNA preparations that are provided because of the efficient sample washing possible in this format.
Multiple samples can be manipulated with a permanent magnet in a microtitre plate format as shown in Figure 10.17. The alternative implementation, also using immobilized
Figure 10.17 Microtitre plate magnetic separator used by Uhlen for automated solid state DNA sequencing.

348 DNA SEQUENCING: CURRENT TACTICS
Figure 10.18 Microtitre plate multipin device used by Landegren for automated DNA sequencing and related automated DNA manipulations.
streptavidin, employs a 48-pin device instead of magnets (Fig. 10.18). Here the immobilized DNA is captured on the ends of plastic pins which have been loaded with strepta- vidin-conjugated microbeads. A very high density of strepavidin can be generated in this
way. It seems clear that by combining magnetic beads and plastic pins, one may be able to automate even more elaborate protocols easily. Recently Landegren reported a very clever variation of this scheme in which the DNA sequencing chemistry is carried out on
a plastic comb of the type used to cast the sample slots in a sequencing gel. The teeth of the comb contained immobilized streptavidin beads. Once the chemistry was completed, the contents of the entire comb were loaded onto a DNA sequencing gel by inserting the comb into a gel with wells containing formamide. This solvent disrupts the binding between streptavidin and biotin, denatures the DNA, and releases the DNA samples into the
gel. Apparently the formamide has no serious deleterious consequences on the subsequent electrophoresis. Thus, in a very simple way, the problem of automated gel loading has effectively been solved.
FUTURE IMPROVEMENTS IN LADDER SEQUENCING
Using all the power of current technology, the very best sequencing laboratories can gen-
erate more than 10 |
5 |
bp of raw DNA |
sequence per day per worker. Lanes read to 600 and |
|
700 bases are common. The entire process |
is fully automated from colony picking |
to |
||
DNA sample preparation to gel loading and running, to the raw sequence analysis and en- |
|
|||
try into a database. Only gel casting and sequence editing are still manual. |
|
|||
A number of different approaches are being tested to see if the throughput of ladder |
||||
sequencing can be further improved. Here we |
will describe some or the more promising |
|||
or more novel attempts. The basic issues are |
how to extend a ladder to longer sizes, how |
|||
to perform the fractionation more rapidly, how |
to increase the number of different sam- |
|||
ples that can be handled simultaneously, and how to read the data more rapidly. A number |
|
|||
of the approaches share the feature that they |
use very thin samples. The advantages |
of |
||
such gels for increasing |
speed |
were described earlier. A disadvantage is that thin |
gels |
|
mean lower amounts of sample, and this requires greater detection sensitivity.
|
|
APPROACHES TO DNA SEQUENCING BY MASS SPECTROMETRY |
349 |
|||
As detectors are improved, it is to be expected that larger numbers of samples will be |
|
|||||
loaded on each gel by using closer spaced and narrower lanes. One limitation with the |
|
|||||
current ALF system is its single color detection; yet the high sensitivity afforded by hav- |
|
|||||
ing the laser in the plane of the gel is a clear advantage. In principle, one could use multi- |
|
|||||
ple lasers in the gel, at different positions, and each could be accompanied by a suitable |
|
|||||
detector array. Ansorge has developed such an instrument, which clearly will have higher |
|
|||||
throughput since each lane will then be available for multicolor sequencing. |
|
|||||
One method of diminishing sample size while retaining sensitive detection is to use a |
|
|||||
fluorescent microscope as the detector. In Chapter 7 the power of confocal scanning laser |
|
|||||
microscopy for FISH was described. This microscope also makes an excellent detector |
|
|||||
for direct scanning of fluorescent-labeled DNA samples in gels. The advantage of the |
|
|||||
confocal microscope is that it gathers emission very efficiently from a very narrow verti- |
|
|||||
cal slice through the sample. Light that emanates from above or below this plane is not |
|
|||||
imaged. Thus the confocal microscope can detect fluorescence from inside a capillary or |
|
|||||
thin slab without background due to |
scattering from the interface between the capillary |
|
||||
and the gel, or the interface between the capillary and the external surroundings. This is a |
|
|||||
major improvement. One consequence is that the capillaries are scanned off line, in order |
|
|||||
to take full advantage of the scanning speed of the microscope. Dense bundles of capillar- |
|
|||||
ies can be made, loaded in parallel |
with multiple headed syringes, run in parallel, and |
|
||||
scanned together (Fig. 10.19). The increase throughput ultimately achievable with this ap- |
|
|||||
proach may be considerable. A potential limitation is the difficulty in making gel-filled |
|
|||||
capillaries. This will be alleviated somewhat as it becomes possible to use liquid (non- |
|
|||||
crosslinked) gels instead of solid (crosslinked) gels. This is because solid gels must be |
|
|||||
polymerized within the capillary, while |
liquid gels can just be poured into the capillary. |
|
||||
The alternative to capillaries is to |
use large thin gel slabs. This simplifies the optics |
|
||||
needed for on-line detection of the DNA. |
In anticipation of the considerable demands |
|
||||
placed on detector systems by fast running, thin gels, a number of alternative new detec- |
|
|||||
tors are being developed as possible readout devices for fluorescence-based DNA se- |
|
|||||
quencing. |
|
|
|
|
|
|
APPROACHES TO |
DNA |
SEQUENCING |
BY |
MASS |
SPECTROMETRY |
|
A separate approach to improving ladder sequencing is to change the way in which the la- |
|
|||||
beled DNA fragments are detected. Here considerable attention has been given to mass |
|
|||||
spectrometry. There are actually three ways in which mass spectrometry might be used, in |
|
|||||
principle, to assist DNA sequencing. In the simplest case the mass spectrometer is used as |
|
|||||
a detector for a mass label attached to the DNA strand in lieu of a fluorescent label. |
|
|||||
Alternatively, the mass of the DNA molecule itself can be measured. In this case the mass |
|
|||||
spectrometer replaces the need for gel |
electrophoresis; it separates the DNA molecules |
|
||||
and detects their sizes. The most ambitious and difficult potential use of mass spectrome- |
|
|||||
try would involve a fragmentation analysis of the DNA and the determination of all of the |
|
|||||
resulting species. In |
this |
way the mass spectrometer would replace all of the chemistry |
|
|||
and electrophoresis steps in conventional ladder sequencing. We are a long way from ac- |
|
|||||
complishing this. In this section we will |
discuss |
each of these potential applications of |
|
|||
mass spectrometry to enhance DNA sequencing. |
|
|
|
|||
Mass spectroscopy is almost as sensitive a detector as fluorescence, with some instru- |
|
|||||
ments having sensitivities |
of the order |
of |
thousands of atoms or molecules, and special |
|
||

350 DNA SEQUENCING: CURRENT TACTICS
(a)
|
Computer |
Amplifier |
> |
|
|
|
|
590 |
|
|
|
Mirror |
|
nm |
|
|
580 |
|
|
|
|
nm |
|
|
|
|
Spatial filter |
555 |
|
|
|
Lens |
|
|
|
|
nm |
|
|
|
|
525 |
Long pass filter |
|
|
|
|
|
|
|
|
nm |
|
|
|
PMT |
|
Dichroic beam splitter |
|
|
Spectral filter |
|
Mirror |
|
|
Laser (488 nm) |
|
Dichroic beam splitter |
|
|
Objective |
|
Detection zone |
|
|
Translation stage |
|
||
|
|
|
|
|
|
Separation |
|
Buffer reservoir |
|
|
capillaries |
|
||
|
|
|
|
|
|
— |
|
+ |
|
|
|
High-voltage |
|
|
|
|
power supply |
|
|
|
|
(b) |
|
|
Figure 10.19 |
Apparatus for DNA sequencing by capillary electrophoresis. |
|
(a ) An array of gel- |
|
filled capillaries |
used for DNA sequencing. ( |
b ) On-line |
detection by |
confocal scanning fluores- |
cence microscopy. Figure provided by Richard Mathies. Figure also appears in color insert.

|
|
|
|
|
APPROACHES TO DNA SEQUENCING BY MASS SPECTROMETRY |
351 |
|||
techniques such as ion cyclotron resonance mass spectrometry having even greater sensi- |
|
|
|||||||
tivity. However, the |
principal |
potential advantages of mass spectra over fluorescence |
is |
|
|||||
that |
isotopic |
labeling |
leads |
to |
much less of a perturbation of electrophoretic properties |
|
|||
than fluorescent labeling, and the number of easily used isotopic labels far exceeds the |
|
||||||||
number of fluorophores that could be used simultaneously. Mass spectrometers actually |
|
|
|||||||
measure the ratio of mass to charge; the best instruments have a mass to charge resolution |
|
|
|||||||
of better than 1 part in a million. Thus asking a mass spectrometer to distinguish between, |
|
||||||||
say, |
two |
isotopes like |
|
|
34S and |
36S is not very demanding if these isotopes reside in small |
|
||
molecules. |
|
|
|
|
|
|
|
||
|
One basic strategy in using mass spectrometry as a DNA sequencing detector simply |
|
|
||||||
replaces |
the |
fluorophore with |
a stable isotope. Two approaches have been explored. In |
|
|||||
one |
case |
four |
different |
stable |
isotopes of sulfur would be used as a 5 |
label |
incorporated, |
||
for |
example, as thiophosphate. In the other |
case a metal chelate is attached at the 5 |
|
-end |
|||||
of the primer, and different stable metal isotopes are used. Some of the possibilities are |
|
||||||||
shown in Table 10.1. Since many of the divalent ions in the table have very similar chem- |
|
|
|||||||
istry, chelates can be built that, in principle, would bind many different elements. Thus, |
|
||||||||
when all the isotopes are considered, there is the possibility of doing analyses with more |
|
|
|||||||
than 30 different colors. Whether sulfur or metal isotopes are used, the sample must be |
|
||||||||
vaporized and the DNA destroyed so that the only mass detected is that of a small mole- |
|
|
|||||||
cule |
or |
single atom containing |
the isotope. With sulfur labeling, one possible role |
for |
|
||||
mass spectrometry is as an on-line detector for capillary electrophoresis. DNA fragments |
|
|
|||||||
are eluted from the capillary into a chamber where the sample is burned, and the resulting |
|
|
|||||||
SO 2 is ionized and detected. |
|
|
|
|
|
||||
|
With metal labeling, a much more complex process is used to analyze the sample by |
|
|
||||||
mass |
spectrometry. This is a |
technique called resonance ionization spectroscopy (RIS), |
|
||||||
and it is illustrated in Figure 10.20. Here mass spectrometry would serve to analyze a fil- |
|
||||||||
ter blot, or a thin gel, directly, off line. In RIS just the top few microns of a sample are ex- |
|
||||||||
amined. Either a strong laser beam or an ion beam is used to vaporize the surface of the |
|
||||||||
sample, creating a mixture of atoms and ions. The beam scans the surface in a raster pat- |
|
|
|||||||
tern. Any ions produced are pulled away by a strong electric field. Then a set of lasers is |
|
||||||||
used |
to |
ionize a particular element of interest; in our case this is the metal atom used |
as |
|
|||||
the label. Because ionization energies are higher than the energy in any single laser pho- |
|
||||||||
ton, two or more lasers must |
be used in tandem to pump the atom up to its ionization |
|
|
||||||
state. Then |
it is detected by |
mass spectrometry. The same set of lasers can be used to |
|
||||||
Figure 10.20 |
Resonance ionization mass spectrometry (RIS). |
(a ) Schematic design of the instru- |
|
ment used to scan |
a surface. ( |
b ) Three electronic states used for |
the resonance ionization of metal |
atoms. |
|
|
|
352 DNA SEQUENCING: CURRENT TACTICS
excite all of the different stable isotopes of a particular element; however different lasers may be required when different elements are to be analyzed.
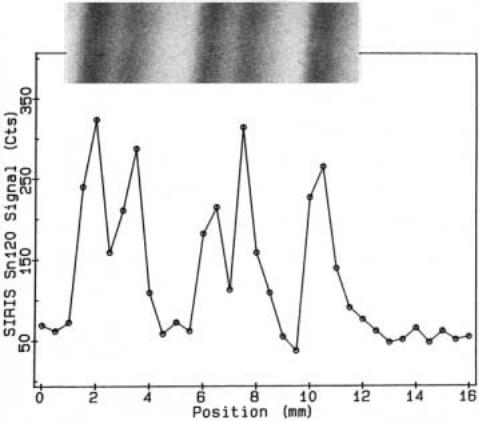
An example of RIS as applied to the reading of a DNA sequencing gel is shown in Figure 10.21. The method clearly works; however, it would be helpful to have higher signal to noise. Actually RIS is an extremely sensitive method, with detection limits of the order of a few hundreds to a few thousands of atoms. Very little background should be expected from most of the particular isotopes listed in Table 10.2, since many of these are not common in nature, and in fact most of the elements involved, with the notable excep-
tion of iron and zinc, are not common in biological materials. The problem is that gel electrophoresis is a bulk fractionation; very few of the DNA molecules separated actually lie in the thin surface layer that can be scanned. Similarly typical blotting membranes are also not really surfaces; DNA molecules penetrate into them for quite a considerable distance.
To assist mass spectrometric analysis of DNA, it would clearly be helpful to have simple, reproducible ways of introducing large numbers of metal atoms into a DNA molecule
and firmly anchoring them there. One approach to this is to synthesize base analogs that have metal chelates attached to them, in a way that does not interfere with their ability to
Figure 10.21 Example of analysis of a DNA sequencing gel lane by RIS. Gel image appears at top; RIS signal below. Provided by Bruce Jacobson. See Jacobson et al. (1990).

|
|
APPROACHES TO DNA SEQUENCING BY MASS SPECTROMETRY |
|
353 |
|||
TABLE 10.2 Stable Metal Isotopes Bound by Metallothionein |
|
|
|
|
|||
|
|
|
|
|
|
|
|
26 |
Fe |
54 |
Fe |
50 |
Sn |
112 |
Sn |
|
56 Fe |
|
114 |
Sn |
|||
|
|
|
|
||||
|
|
57 Fe |
|
|
115 |
Sn |
|
|
|
58 Fe |
|
|
116 |
Sn |
|
|
|
|
|
|
|
117 |
Sn |
|
Co |
56 Co |
|
|
118 |
Sn |
|
27 |
|
|
|
|
119 |
Sn |
|
|
|
|
|
|
|
||
|
Ni |
58 Ni |
|
|
120 |
Sn |
|
28 |
60 Ni |
|
|
122 Sn |
|||
|
|
|
|
||||
|
|
61 Ni |
|
|
124 |
Sn |
|
|
|
62Ni |
|
|
|
|
|
|
|
64 Ni |
|
Au |
197 Au |
||
|
|
|
|
79 |
|
|
|
29Cu |
63Cu |
80 Hg |
196 Hg |
||||
|
|
65 Cu |
|
|
198 Hg |
||
|
|
|
|
|
|
199 Hg |
|
30 |
Zn |
64 Zn |
|
|
200 |
Hg |
|
|
66 Zn |
|
|
201 |
Hg |
||
|
|
|
|
||||
|
|
67Zn |
|
|
202 Hg |
||
|
|
68Zn |
|
|
204 |
Hg |
|
|
|
70 Zn |
|
|
|
|
|
|
|
|
|
|
Pb |
204 |
Pb |
|
Ag |
107 Ag |
82 |
206 |
Pb |
||
47 |
|
|
|||||
|
109 Ag |
|
|
207 Pb |
|||
|
|
|
|
||||
|
|
|
|
|
|
208 Pb |
|
48 |
Cd |
106 Cd |
|
|
|
|
|
|
108 Cd |
|
Bi |
209 |
Bi |
||
|
|
|
|||||
|
|
110 |
Cd |
83 |
|
|
|
|
|
|
|
|
|
||
|
|
111 |
Cd |
|
|
|
|
|
|
112 Cd |
|
|
|
|
|
|
|
113 Cd |
|
|
|
|
|
|
|
114 |
Cd |
|
|
|
|
116 Cd
Total 50 species
hybridize to complementary sequences. An example is shown in Figure 10.22. An alternative approach is to adapt the streptavidin-biotin system to place metals wherever in a DNA one places a biotin. This can be done by using the chimeric fusion protein shown in
Figure 10.23. This fusion combines the streptavidin moiety with metallothionein, a small cysteine-rich protein that can bind 8 divalent metals or up to 12 heavy univalent metals. The list of different elements known to bind tightly to metallothionein is quite extensive. All of the isotopes in Table 10.2 are from elements that bind to metallothionein. The fusion protein is a tetramer because its quaternary structure is dominated by the extremely stable streptavidin tetramer. Thus there are four metallothioneins in the complex, and each retains its full metal binding ability. As a result, when this fusion protein is used to label biotinylated DNA, one can place 28 to 48 metals at the site of each biotin. The use of this fusion protein should provide a very substantial increase in the sensitivity of RIS for DNA detection.

354 DNA SEQUENCING: CURRENT TACTICS
Figure 10.22 A metal chelate derivative of a DNA base suitable as an RIS label.
While mass spectrometry has great potential to detect metal labels in biological systems, a drawback of the method is that current RIS instrumentation is quite costly. Another limitation is that RIS destroys the surface of the sample, so it may be difficult to
read each gel or blot more than once. Alternative schemes for the |
use of |
metals as |
labels |
in DNA sequencing exist. One is described in Box 10.1 |
|
|
|
The second way to use mass spectrometry to analyze DNA sequences ladders is to at- |
|||
tempt to place the DNA molecules that constitute the sequencing |
ladder |
into the |
vapor |
phase and detect their masses. In essence this is DNA electrophoresis in vacuum. A key requirement of the approach is to minimize fragmentation once the molecules have been placed in vacuum, since all of the desired fragmentation needed to read the sequence has already been carried out through prior Sanger chemistry; any additional fragmentation is
confusing and leads to a loss |
in |
experimental sensitivity. Two |
methods show great |
promise for placing macromolecules |
into |
the gas phase. In one, called |
electrospray (ES), |
a fine mist of macromolecular solution is sprayed into a vacuum chamber; the solvent evaporates and is pumped away. The macromolecule retains an intrinsic charge and can
be accelerated by electrical fields, and its mass subsequently measured. In the second approach, matrix-assisted laser desorption and ionization (MALDI), the macromolecule is suspended in a matrix that can be disintegrated by light absorption. After excitation with a pulsed laser, the macromolecule finds itself suspended in the vapor phase; it can be accel-
erated |
if |
it has |
a charge. These two procedures |
appear |
to work very well for proteins. |
They |
also |
work |
very well on oligonucleotides, |
and a |
few examples of high-resolution |
mass spectra have been reported for compounds with as many as 100 nucleotides.
Figure 10.23 Structure of a streptavidin-metallothionein chimeric protein capable of bringing 28 to 48 metal atoms to a single biotin-labeled site in a DNA.