

11. Thermochemistry of olefins, carbonyl compounds and imines |
557 |
−17.1± 2.6 |
−6.8 ± 2.6 |
− 10.3 ± 2.7
SCHEME 3
trans-3-heptene to trans-2-methyl-3-hexene ( 6.7 š 1.2 kJ mol 1) and for trans-3-octene to trans-2,2-dimethyl-3-hexene ( 14.9 š 2.2 kJ mol 1). When the butyl groups are substituted on a double bond which is cis to a methyl group (Scheme 4), the steric effect on the enthalpy is evident upon isomerization to the tertiary group, again using the mean value for 4,4-dimethyl-2-pentene21,28. Enthalpies of isomerization of n-Pr to i-Pr in cis-CH3CHDCHPr and the cis-CH3CH2CHDCHPr are 9.9 š 2.1 kJ mol 1 (using the mean value for the i-Pr-substituted compound16,26) and 7.5 š 2.0 kJ mol 1 respectively, while that for butyl rearrangement in cis-3-octene to cis-5,5-dimethyl-3- hexene is C1.5 š 3.2 kJ mol 1.
−4.9 ± 2.5 |
−6.4 ± 2.3 |
+1.5 ± 2.8
SCHEME 4
Unfortunately, the data are sparse for 1,1-disubstituted ethenes, and so the few values we have do not appear in a Scheme. Complicating the analysis are the multiple enthalpies of formation for 2,3-dimethyl-1-butene16,26,32, 2,3,3-trimethyl-1-butene21,27 and 2-ethyl- 3-methyl-1-butene16,28 where the extreme values in a set are not within each other’s uncertainty intervals. We would like to compare these isomerizations with those of the corresponding cis-1,2-disubstituted ethenes since they are relevant to the discussion of steric effects in disubstituted ethenes, Because we have no other way of gauging the magnitude of these effects, we report the enthalpy of isomerization ranges calculated using the extreme enthalpies of formation: H2CDC CH3 n-Pr ! H2CDC CH3 i -Pr, 3.5š
1.6 to 7.4 š 2.3 kJ mol 1; H2CDC CH3 n-Bu ! H2CDC CH3 t-Bu, 6.8 š 1.8 to 11.4 š 1.8 kJ mol 1; and H2CDC CH2CH3 n-Pr ! H2CDC CH2CH3 i -Pr,
2.8 š 2.6 to 8.2 š 3.0 kJ mol 1. Regardless of which enthalpy of formation is used, it seems that isomerization to the tertiary substituent is more favourable in the 1,1- disubstituted ethenes than in the cis-1,2-disubstituted ethenes. We will consider again these enthalpy-of-formation values in the context of ketone isomerizations in a later section.

558 |
Suzanne W. Slayden and Joel F. Liebman |
The only reported enthalpies of formation for 2-methyl-1-heptene29 and 2-methyl-3- ethyl-1-pentene16,21 are identical: 100.3 kJ mol 1. On the one hand, we might expect at least some stabilization due to secondary branching effects as was found for the n- Pr to i-Pr example cited above. On the other hand, if the actual stabilization is at the lower end of the cited ranges for the propyl isomerizations, it may be vanishingly small for the pentyl isomerization. That is, the enthalpy of isomerization might decrease as nc increases for these two homologous series. Lastly, the n-pentyl to neopentyl isomerization enthalpy, from 2-methyl-1-heptene to 2,4,4-trimethyl-1-pentene, is 10.6 kJ mol 1, about half the value found for the corresponding isomerization in mono-substituted alkenes (from 1-heptene to 4,4-dimethyl-1-pentene21,28 it is ca 19 kJ mol 1).
III.CYCLIC OLEFINS WITH A −CH=CH− GROUP
A.Cis-Cycloalkenes
In this section we consider the energetics of molecules with the generic formula cis- or (Z)-cyclo-[(CH2)n 2 (CHDCH)] (1). A natural comparison of the enthalpies of formation of these olefins is with their saturated analogs, the cycloalkanes, with the generic formula, cyclo-[(CH2)n 2 (CH2CH2)] [i.e. (CH2)n, 2]. Using equation 13, let us now define the difference quantity υ13(n) as the difference of the gas-phase enthalpies of formation of the cycloalkene of interest and the related cycloalkane:
υ13 n H°f cyclo-[ CH2 n], g H°f cyclo-[ CH2 n 2 CHDCH ], g 13
(CH2 )n−2 (CH2 )n−2
(1)(2)
Throughout our thermochemical studies, we have employed numerous difference quantities. Many have been conceptual conveniences to aid us in predicting or explaining a number of interest. Some of these difference quantities correspond to the energetics of a real reaction. In that H°f (H2, g) is defined to be precisely equal to zero, the quantity υ13(n) may be recognized as numerically equalling the enthalpy of hydrogenation of the n-membered ring olefin. The numerical values of the enthalpies of formation of the various cycloalkenes and the corresponding υ13(n) values are found in Table 3. The values of υ13(n) almost all lie between 85 and 130 kJ mol 1, an admittedly large range. The sole exception to that is the n D 3 case, cyclopropene with υ13 D 224 kJ mol 1. This marked deviance is consistent with the ca 50 kJ mol 1 additional destabilization for each trigonal or sp2 carbon that is introduced into a three-membered ring52. Other than that, it is difficult to see a pattern in terms of the ring size or parameter n. Cyclobutene has the next highest value of υ13(n) as befits the high strain of the cycloalkane. The n D 5 and 7 species have nearly identical and much smaller values of strain energy in the cycloalkane53 and nearly identical and much smaller values of υ13(n). Cyclohexane shows an additional decrease in strain energy relative to the other cycloalkanes but the value of υ13 increases! Smaller values of υ13(n) are found for n 8 but no pattern is apparent. From the numerous measurements reported by Roth and coworkers54 for the n D 8 case, we may expect only small revisions of the values of υ13(n) for the intermediate-size 9-,

11. Thermochemistry of olefins, carbonyl compounds and imines |
559 |
|||||
|
TABLE 3. Enthalpies of |
formation of gaseous (Z)- |
|
|||
|
cycloalkenes, cyclo-[(CH2)n 2(CHDCH)], and the corre- |
|
||||
|
sponding values of υ13 n |
|
|
|
|
|
|
n |
Hf° (g) |
Reference |
υ13 n |
|
|
3 |
277.1 š 2.5 |
a |
223.8 š 2.6 |
|
||
a |
|
|||||
4 |
156.7 š 1.5 |
a |
128.3 š 1.6 |
|
||
5 |
33.9 š 1.4 |
a |
110.3 š 1.6 |
|
||
6 |
5.0 š 0.7 |
a |
118.4 š 1.1 |
|
||
7 |
9.2 š 1.1 |
b |
108.9 š 1.5 |
|
||
8 |
28.0 š 1.2 |
c |
96.4 š 0.7 |
|
||
9 |
34.1 |
c |
98.7 |
|
|
|
10 |
67.7 |
c |
86.6 |
|
|
|
12 |
120.2 |
|
110.0 |
|
|
|
a These values are taken from Pedley.
b This value is that found by hydrogenating the cycloalkene in a nonpolar hydrocarbon solvent, see Rogers and coworkers51
c These values are those found by hydrogenating the cycloalkene in glacial AcOH, see Jensen22.
10and 12-membered rings upon correction of the experimentally measured enthalpies of hydrogenation in glacial AcOH.
B. Trans-Cycloalkenes
In this section we consider the energetics of molecules with the generic formula trans- or (E)-cyclo-[(CH2)n 2 (CHDCH)], 3. Again, a natural comparison of the enthalpies of formation is with their saturated analogs, the cycloalkanes, cyclo-[(CH2)n 2 (CH2CH2)] [i.e. (CH2)n]. However, we think that an even simpler comparison is with their respective cis- or (Z)-cyclo-[(CH2)n 2(CHDCH)] isomers because solvent effects on the hydrogenation enthalpies that interrelate cycloalkenes and cycloalkanes should more completely cancel when comparing isomeric cycloalkenes. For ‘small enough’ ring sizes or values of n, the (E)-isomer should be less stable than the (Z)-isomer because of the necessity of twisting the double bond in the former. For ‘large enough’ rings or values of n, the (E)-isomer should be more stable than the (Z)-isomer much as it is found that (E)-acyclic olefins with the CHDCH substructure are more stable than their (Z)-counterparts. The smallest value of n for which both isomeric cycloalkenes have been thermochemically investigated is n D 8 for which the (Z)-isomer is the more stable by ca 47 kJ mol 1 if the results either from hydrogenation calorimetry in the gas phase or a hydrocarbon solvent are employed22,51,54. The less fundamental methodology of hydrogenation calorimetry in glacial AcOH22 suggests a difference of only 36 kJ mol 1. In the absence of thermochemical data from experiments in the former, more innocuous media, from results in the latter22 we find that the difference has narrowed to 12 kJ mol 1 for n D 9, increased to 14 kJ mol 1 for n D 10 and fallen to 2 kJ mol 1 for n D 12. There is no use in
(CH2 )n−2
(3)
560 |
Suzanne W. Slayden and Joel F. Liebman |
comparing the n D 9 and 10 values given the uncertainty in differential solvation effects, but it is safe to assume that the (E)-isomer is more stable for n > 12 species. By contrast, it is safe to assume that n becoming ever smaller will increasingly favour the (Z)-over its (E)-isomer55.
C. Bicycloalkenes
All of the compounds of interest in this section can be described as cycloalkenes with two carbons joined by a polymethylene bridge. We can attempt to understand these species by varying the size of the cycloalkene and/or the length of the bridge, and acknowledge that occasionally there exists the possibility of varying the positions on which this bridge is affixed. Homoconjugatively derived stabilization or destabilization are distinct possibilities and so provide interesting complications7,54. We now consider bicyclo[n.1.0]alkenes by definition of the current class of compounds, the double bond is in the larger ring.
One probe is the difference of the enthalpies of formation of the bicyclo[n.1.0]alk-2- ene, (4), and the related cycloalkene where the affixed CH2 or monomethylene bridge has been replaced by two ‘ H’. For n D 2, 3 and 4, the differences are 17754, 12456 and 12656 kJ mol 1, respectively (bicycloalkene data from References 54 and 56, cycloalkene data from Pedley). Not surprisingly, it is the species with the 4-membered ring (n D 2) that shows a significant difference, in this case marked destabilization suggestive of homoantiaromaticity7,54,56 as well as ‘superstrain’57 in the bicyclopentene arising from the fusion of the two, individually highly strained, rings.
(CH2 )n
(CH2 )n−2
(4)(5)
Now let us turn to the category where the length of the bridge is varied and consider the bicyclo[n.2.0]alkenes that are formally constructed from cyclobutene and a polymethylene bridge attached to C3 and C4 of that cyclobutene, cf. 5. A logical comparison58 entails the difference between the enthalpies of formation of these olefins with the corresponding cycloalkanes with n D 2 atoms in the ring, i.e. those species in which the ‘ CHDCH ’ has been replaced by two ‘ H’. For example, bicyclo[2.1.0]pent-2-ene that was formed from cyclobutene and a one-carbon chain is compared with cyclopropane by loss of the two-carbon CHDCH unit found in all of the bicycloalkenes of the current category. For n D 154, 259, 354 and 454, the derived differences are 281, 233, 216 and 245 kJ mol 1, respectively. While we acknowledge that the compound with n D 1 is again an outlier because of homoantiaromatic as well as superstrain derived destabilization, it is nontheless disconcerting how much the differences for the other bicycles fluctuate. For example, it is not obvious why the n D 3 bicyclic olefin is so stable. Alternatively, one may interrelate the bicyclo[n.2.0]alkene with the cycloalkene formed by replacing the bridgehead C C bond by two C H bonds. For n D 154, 259, 354 and 454, the derived differences are 299, 266, 148 and 147 kJ mol 1. No pattern is found. In the absence of any strain or electronic effect of the double bond, one is replacing a ring of n C 2 atoms by one of n C 4 atoms. Strain in alicyclic hydrocarbons is not monotonic with the number of atoms53,57. As such the near-equality for the above n D 3 and 4 cases is not unlike the situation for the near-equality of the strain energies of 3- and 4-membered cycloalkanes: both appear
11. Thermochemistry of olefins, carbonyl compounds and imines |
561 |
to be numerical ‘accidents’. We may also vary the position of the double bond within the larger ring of the bicycloalkene. Examples are rare but it is unequivocally found with the case of the isomeric norcarenes (i.e. syn-bicyclo[4.1.0]hept-2- and -3-enes 6a and 6b, respectively). The difference as given by Roth and coworkers54 is under one kJ mol 1. However, we hesitate to generalize this near-constancy since these species are formally related to the isomeric 1,3- and 1,4-cyclohexadienes (7a and 7b, respectively) for which the difference in their enthalpies of formation is still disconcertingly contentious8. It is quite clear, however, that anti fusion is destabilizing: from Roth and coworkers54 we find that the anti -bicylo[4.1.0]hept-3-ene (8) is ca 100 kJ mol 1 less stable than its syn-isomer.
(6a) |
(6b) |
(7a) |
(CH2 )n
(7b) |
(8) |
(9) |
In the absence of 1-carbon bridges, the possible complications of homoantiaromatic interactions are strongly minimized and thermochemical analyses would be thought simpler. An example of the effects of varying the bridge length is found with the bicyclo[n.2.2]alkenes (9) wherein the double bond is assumed to be on one of the two bridges. Again let us take the difference of the enthalpies of formation of these species with a ‘ CHDCH ’ group and the species in which this group has been replaced by two ‘ H’, e.g. bicyclo[2.2.2]oct-2-ene is compared with cyclohexane. For n D 059, 160, 261 and 354, the differences are 233, 166, 158 and 115 kJ mol 1. The absence of near-constancy is not surprising: after all, the double bond being replaced is part of a 4-, 5-, 6- and 7-membered ring that is being dismembered. Nonetheless, we may argue again that the n D 0 species is homoantiaromatic, superstrained and therefore an outlier, and so the remaining three values are not that dissonant.
What about hydrogenation of the double bond to form the bicyclo[n.2.2]alkane? The respective enthalpies are 13659, 14260, 13461 and 11562 kJ mol 1. The absence of near-constancy or uniform strain result is found here, although the spread of only ca 30 kJ mol 1 is quite small. The lack of uniformity is again not particularly surprising because enthalpies of hydrogenation for the simple monocyclic cycloalkenes to the saturated cycloalkane are not particularly constant either. (Recall the content of Table 3.) Furthermore, the geometries of monocyclic cycloalkene and cycloalkane rings are different from those found in their bicyclic counterparts. For example, both bicyclo[2.2.2]octene and bicyclo[2.2.2]octane ‘contain’ boat 6-membered rings, a conformation found in neither cyclohexene nor cyclohexane as free monocycles.
562 |
Suzanne W. Slayden and Joel F. Liebman |
D. Polycycloalkenes
There are available measured enthalpies of formation of surprisingly few species and so attempts to find patterns are even more difficult here than before. Even if we consider the simpler quantity of the enthalpy of hydrogenation, we find that few polycyclic species qualify. To provide some semblance of conceptual unity, let us limit our attention to those polycyclic olefins in which the double bond being hydrogenated is found in a 5- or 6-membered ring. As such, we will compare the enthalpies of their hydrogenation with that of the normal, relatively strainless cyclopentene and cyclohexene, with υ13 equalling 110.3 š 1.6 and 118.4 š 1.1 kJ mol 1, respectively.
(10) |
(11) |
(12) |
(13a) |
(13b) |
(13c) |
(14) |
(15) |
(16a) |
(16b) |
1
2
3
4
(17)
11. Thermochemistry of olefins, carbonyl compounds and imines |
563 |
We commence with cyclopentene derivatives. Syn-bicyclo[3.3.0]oct-2-ene (dihydrobiquinacene, 10) has nearly the same enthalpy63 of hydrogenation, 112.1š1.3 kJ mol 1. In order of increasing number of complexity (i.e. increasing number of rings, and if constant, then increasing number of carbons), consider benzvalene (11), tetrahydrotriquinacene (12), three singly unsaturated derivatives of sesquinorbornene (13a, 13b, 13c) and tetrahydrohexaquinacene (14). We find their hydrogenation enthalpies are 13556, 11564, 11354, 15654, 14054 and 10363 kJ mol 1, respectively. Besides these gas-phase numbers, we also may derive the enthalpy of hydrogenation of 3a,4,5,6,7,7a-hexahydro-4,7-methanoindene (15) by taking the difference of Pedley’s cited standard enthalpy of formation and that of the octahydromethanoindene. As solids, the υ13 value of interest is 89.0 š 6.5 kJ mol 1. Related examples are the so-called dihydro-˛- and -ˇ-tricyclopentadiene (16a, 16b) to form the corresponding tetrahydro-˛- and -ˇ-tricyclopentadiene65, respectively. Deriving the enthalpies of hydrogenation by taking the differences of enthalpies of formation of the corresponding solid dihydro and tetrahydro derivatives, the desired υ13 are found to be 101 and 107 kJ mol 1. None of these numbers is particularly useful because of the medium and state of the compound being studied.
There are few cyclohexene derivatives with which to make comparison. Among them are cholest-1-ene (17) and its isomers, cholest-2- and -3-ene for which enthalpies of hydrogenation in glacial AcOH have been reported22 to be 114, 108 and 119 kJ mol 1, respectively. We admit that saturated polycyclic species have complexities of their own in their interplay of structure and energetics. Nonetheless, it would appear that polycyclic analogs of relatively normal olefins have additional, and likewise disconcertingly complex, thermochemical features.
IV. EXO- VS ENDO-CYCLIC DOUBLE BONDS
A. Isomeric Methylcycloalkenes and Methylenecycloalkanes
In this section we compare the enthalpy of formation of isomeric pairs of cyclic olefins, one with an exocyclic double bond and the other with the double bond endocyclic. We start with 1-methylcycloalkenes and related methylenecycloalkanes, species generically 18a and 18b, respectively. From prior experience with cyclopropanes and the thermochemical consequences of replacement of sp3 carbons by sp2 carbons in three-membered rings52, we expect 1-methylcyclopropene to be considerably less stable than methylenecyclopropane because the former compound has two trigonal carbons within the ring while the latter has but one. And so it is found56: the former has a gas-phase enthalpy of formation 43.1 š 2.2 kJ mol 1 more positive than the latter.
(CH2 )n −2 |
(CH2 )n −2 |
(18a) |
(18b) |
Our enthalpy of formation archive lacks data for the corresponding cyclobutene/cyclobutane derivatives. Nonetheless, the enthalpy of hydrogenation22 of both 1-methyl- cyclobutene and methylenecyclobutane in the same solvent (AcOH) has been reported. Because the same product, methylcyclobutane, is formed, it is not unreasonable to assume that the difference of the enthalpies of hydrogenation of the two olefins roughly equals the difference of the enthalpies of formation of the olefins66. It is thus found that the
564 Suzanne W. Slayden and Joel F. Liebman
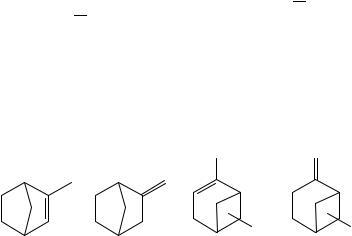
exocyclic olefin is less stable than the endocyclic by only 4 kJ mol 1. The hydrogenation enthalpy difference of 1-methyl-cyclopentene and methylenecyclopentane, favouring the former, is increased to 16.3 and 14.6 kJ mol 1 in glacial acetic acid and the innocuous hexane67, respectively. These values are essentially indistinguishable from the differences of 15.8 š 1.3 and 16.3 š 1.1 kJ mol 1, respectively, for the archival values of enthalpies of formation (derived from combustion calorimetry) for the gas phase and neat liquids, respectively. However, before we become too confident or merely too complacent, the difference is seen to decrease to 9.6 kJ mol 1 for that between 1-methylcyclohexene and methylenecyclohexane as determined by hydrogenation measurements (again in AcOH) but is 18.1 š 3.9 and 19.9 š 3.9 kJ mol 1 for the gaseous and neat liquids as determined by combustion calorimetry. For the cycloheptane case we only have hydrogenation data which suggest that the endocyclic species is less stable than its exo isomer by 9.6 kJ mol 1.
B. Related Polycyclic Species
Can we generalize these results? Admitting the seeming exception of cyclobutene results, it would appear that the endocyclic olefin is more stable by ca 15š5 kJ mol 1 than its exocyclic isomer: even the cyclopropane species obeys this conclusion if we remember to correct for the ca 50 kJ mol 1 destabilization in the 1-methylcyclopropene because of the second sp2 carbon52. We now ask about bicyclic olefins. Data are quite sparse here. One example is 2-methylbicyclo[2.2.1]hept-2-ene and 2-methylenebicyclo[2.2.1]heptane, species 19a and 19b, respectively. Admittedly for the liquid phase and determined by enthalpy of combustion we find the endocyclic isomer is less stable than the exocyclic isomer by 8.6 š 2.2 kJ mol 1. By contrast, the enthalpies of formation of the isomeric bicyclo[2.2.2]octane derivatives favour the endocyclic isomer and differ by 7.5 š 3.3 and 9.4 š 3.9 kJ mol 1 for the liquid and gaseous phase, respectively, consonant with the relative stability of the monocyclic olefins. Relatedly, ˛- and ˇ-pinene, the multiply methylated bicyclo[3.1.1]heptenes 20a and 20b respectively, have enthalpies of formation which differ by 8.7 š 3.7 and 10.4 š 4.1 kJ mol 1 favouring the endocyclic isomer for the liquid and gaseous states, respectively. We have no explanation for the discrepancy for the bicyclo[2.2.1]heptane derivatives68.
(19a) |
(19b) |
(20a) |
(20b) |
C. Isomeric Ethylcycloalkenes, Ethylidenecycloalkanes
and Vinylcycloalkanes
What about the comparison of isomeric 1-ethylcycloalkenes and ethylidenecycloalkanes, generically 21a and 21b respectively? This is perhaps a ‘fairer’ competition because both olefins are internal, i.e. neither has a DCH2 group, and so it is expected that the enthalpies of formation will be closer. For the isomeric cyclopentane derivatives, the ‘idene’ isomer is more stable by but 1.6 š 1.3 kJ mol 1. For the isomeric olefins with a 6-membered ring, the difference is 1.9 š 1.3 kJ mol 1.
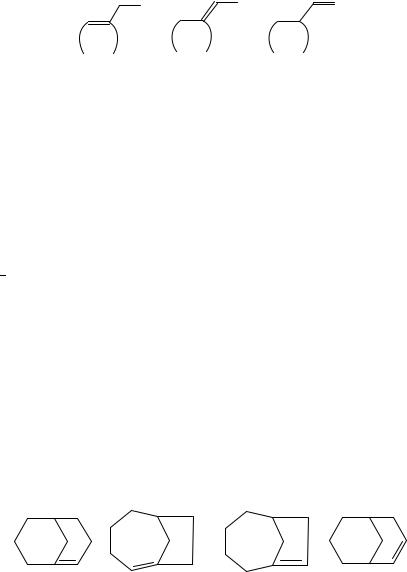
11. Thermochemistry of olefins, carbonyl compounds and imines |
565 |
(CH2 )n − 2 |
(CH2 )n − 2 |
(CH2 )n − 2 |
(21a) |
(21b) |
(21c) |
The appendage of a two-carbon chain on a ring allows for discussion of one more isomeric type of olefin that has the same carbon framework, vinylcycloalkanes here labelled as 21c. For the ethylidenecyclopentane/vinylcyclopentane comparison, the liquid of the former is more stable than that of the latter by 23.5 š 1.4 kJ mol 1. The enthalpies of vapourization of the two isomers should be nearly identical and so the gas-phase difference should be nearly the same. The ethylidenecyclohexane/vinylcyclohexane comparison can be made in both the liquid and gaseous phases. As liquid, the former is more stable by 14.8 š 1.1 kJ mol 1, while as gas, by 12.6 š 1.2 kJ mol 1. It is not obvious why the ‘idene’/vinyl comparison is so different for the two different ring sizes. However, whether cyclopropane mimics cyclopentane or cyclohexane, prior experience on the effects of sp2 carbons in three-membered rings52 suggests that ethylidenecyclopropane will have a ca 50 kJ mol 1 destabilization relative to the other rings. There should thus be a ca 30 40 kJ mol 1 difference between vinylidenecyclopropane and vinylcyclopropane with the latter more stable. The difference derived from taking the enthalpies of formation of the two species suggested in Reference 56 is 34 kJ mol 1, in fine agreement.
V. BREDT’S RULE, BRIDGEHEAD OLEFINS AND CYCLIC SPECIES
WITH A >C=CH− OLEFINIC LINKAGE
For somewhat over seventy years, the structural proscription against bridgehead double bonds known as Bredt’s rule has provided a guiding principle for strained molecules. More precisely, it is suggested that bicyclo[m.n.p]alk-1-enes are destabilized for small, but nonzero, m, n and p. The enthalpy of formation of three such species is available54 from hydrogenation calorimetry. These are the isomeric bicyclo[3.3.1]non-1-ene (22a), bicyclo[4.2.1]non-1-ene (22b) and bicyclo[4.2.1]non-1(8)-ene (22c), bicyclo[4.2.1]non-1,8-ene) with gas-phase enthalpies of formation of 31, 74 and 50 kJ mol 1, respectively. These three numbers alone inadequately address the issue of bridgehead destabilization because the alicyclic carbon skeleton is not the same for all three olefins.
(22a) |
(22b) |
(22c) |
(22d) |
The first species may be compared with the ‘normal’ (i.e. non-bridgehead) olefin bicyclo[3.3.1]non-2-ene, 22d. The former is less stable than the latter by some 80 kJ mol 1 even though the second species has a CHDCH linkage and the first has a >CDCH linkage. (For calibration, one may compare the relative stability of the hardly strained monocyclic olefins; our primary archive shows that of the isomeric 3- and 1- methylcyclopentenes, the latter is more stable69 than the former by 11.2 š 1.0 kJ mol 1.)
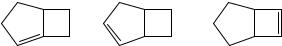
566 Suzanne W. Slayden and Joel F. Liebman
The ca 30 š 10 kJ mol 1 higher enthalpy of formation of the bicyclo[4.2.1]non-1- enes over the bicyclo[3.3.1]non-1-ene should not be totally ascribed to differences in non-planarity, either pyramidization or twisting, or to any other distortion of the olefins. After all, molecular mechanical calculations reported elsewhere54 show that the saturated bicyclo[4.2.1]nonane has a higher enthalpy of formation than bicyclo[3.3.1]nonane by 25 kJ mol 1. As such, much of the difference between the three bicyclononenes is found in the saturated compound, and so documents the importance of the quantitative concept of olefin strain (also called OS or OSE), the difference of strain energy of the olefin and its hydrogenated counterpart70,71. Yet neither this new strain energy definition, per se, or anything else known to the authors allows us to understand the relative enthalpies of formation of the isomeric bicyclo[4.2.1]nonenes. As such, we reluctantly reaffirm, rather than answer, the enigma posed earlier by Warner72 in a recent review on Bredt’s rule and its violations.
What about the case in which the double bond contains one carbon on a 0-length bridge? Such species are often considered as violating Bredt’s rule. We can make but one thermochemically meaningful comparison of such a species with any other olefin that has the same alicyclic framework but lacks this Bredt-violating, strain-increasing feature. This is for bicyclo[3.2.0]hept-1-ene (23a) in comparison to its isomers, the -2- and -6-ene (23b and 23c): 23a is 49 and 28 kJ mol 1 less stable than 23b and 23c, respectively.
(23a) |
(23b) |
(23c) |
What about the case in which both carbons of the double bond are on the ‘0’ bridge? As these species lack the >CDCH olefinic linkage that defines the key compounds of this section, such compounds are deferred elsewhere in this chapter. We merely note at this juncture that they generally have extra strain for those species in which the remaining bridges are short.
VI. CYCLIC OLEFINS CONTAINING A TETRASUBSTITUTED DOUBLE BOND
A. Monocyclic 1,2-Disubstituted Cycloalkenes
The primary examples of this class of compounds are the 1,2-dialkylcycloalkenes (24: n, R1, R2). The simplest thermochemical comparison we can make is with the corresponding unsubstituted cycloalkene, i.e. R1 D R2 = H. A suitable probe of the effect of the dialkylation is the enthalpy of the formal transalkylation reaction
cycloCH2 n 2 RCDCR C C2H4 !cycloCH2 n 2 HCDCH
C Z RCHDCHR |
(14) |
Small thermochemical effects of dialkylation are expected. That is, the above reaction is expected to be essentially thermoneutral because both steric and electronic differences between reactants and products are expected to be minimal. Indeed, this is found for the analogous acyclic reaction
Me2CDCMe2 C C2H4 !Z-Me2 CHDCH C Z-MeCHDCHMe
D 2[Z-MeCHDCHMe] |
(15) |