

Влияние межъядерного расстояния
Энергии всех состояний молекулы зависят от расстояния между ядрами, которое входит в выражения для кулоновского и обменного интегралов, а также интеграла перекрывания.

Зависимость величины кулоновского (J) и обменного (K) интегралов от межъядерного расстояния в молекуле водорода
Проведя множество расчетов для серии межъядерных расстояний, можно построить зависимость E=f(rab), график которой имеет следующий вид:
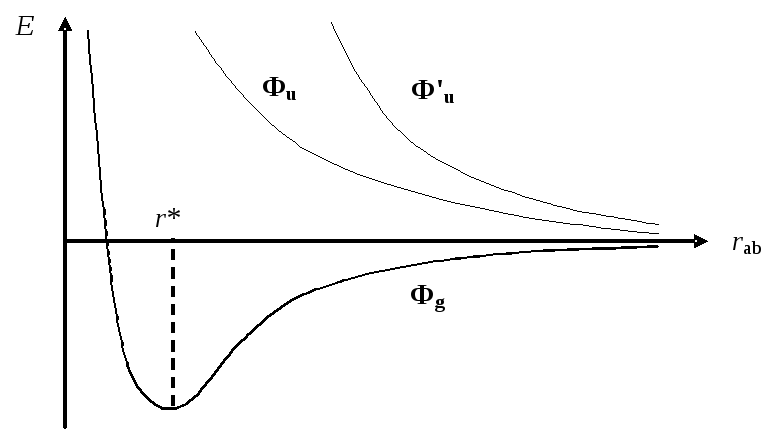
Из графика ясно видно, что процесс сближения двух атомов может приводить к двум принципиально разным результатам. Первый (Фg) — возникновение сил притяжения,что сопровождается уменьшением полной энергии системы по мере сближения атомов. Второй (ФuиФ'u) — возникновение сил отталкивания,что сопровождается возрастанием полной энергии системы. Ясно, что в первом случае сближение атомов будет происходить самопроизвольно до достижения состояния устойчивого равновесия с минимальной энергией (rab=r*).
Именно этот случай соответствует образованию химической связи между атомами. Полезно также обратить внимание на то, что образование химической связи сопровождается спариванием электронов атомов (состояние Фg синглетное), тогда как возникновение сил отталкивания наблюдается для триплетного состоянияФu, когда электроны не спарены. Можно сказать, что спаривание валентных электронов является необходимым признаком химического взаимодействия атомов.
Этот признак, однако, недостаточен, поскольку в состоянии Ф'uэлектроны спарены, но химического взаимодействия не наблюдается. Причина такого различия заключается в различной форме электронного облака. Если сравнить пространственные части двух синглетных волновых функций:
Фg=AB+BAиФ'u=AA–BB
то обнаружится, что во втором случае в электронном облаке имеется разрыв (узловая поверхность, проходящая между ядрами):

Следовательно, для образования химической связи кроме спаривания необходимо еще одно условие — надлежащее распределение электронной плотности в пространстве.
Метод мо
В этом методе молекула рассматривается в рамках орбитальной модели, точно так же, как многоэлектронный атом, только способы движения электронов описываются не атомными, а молекулярными орбиталями(МО). Молекулярная орбиталь отличается тем, что она является многоцентровой,т.е. охватывает сразу несколько атомных ядер.
Глобальная волновая функция молекулы в методе МО строится в виде определителя Слэтера, каждая строка которого включает все занятые электронами МО. Для этого, однако, необходимо знать явный вид МО. Эта задача решается на основе принципа суперпозиции — неизвестная функция заменяется линейной комбинацией известных базисных функций:
i=Ci11+Ci22+ …
Для построения таких комбинаций в методе используется атомный базис, аналогичный тому, который применяется в методе ВС. Единственная разница заключается в том, что в методе ВС анализируется глобальная функция молекулы, а в методе МО — одноэлектронные молекулярные орбитали. В методе ВС с помощью перегородок разрезается многоэлектронное облако, включающее все электроны молекулы. В методе МО тем же набором перегородок разрезается одноэлектронное облако. Поэтому здесь в качестве резонансных форм выступает единственный электрон, локализованный в окрестности одного из ядер. Его движение в этом случае описывается соответствующей атомной орбиталью. В результате, каждая МО представляется в виде линейной комбинации базисного набора атомных орбиталей (ЛКАО).
Основная проблема метода МО сводится к вычислению коэффициентов разложения для каждой из МО. При решении этой задачи следует учитывать два важных условия.
1) ортонормированность:ij dv=ij(1 приi=jи 0 приij),
2) пространственная симметрия:iНП ТГС (всякая МО должна описываться одним из типов симметрии точечной группы молекулы).
Для молекулы водорода принято классифицировать МО относительно операции инверсии — на четные,для которыхi(g) = (+1)(g) инечетные,для которыхi(u) = (–1)(u).
Перейдем к рассмотрению молекулы водорода. Для нее атомный базис состоит всего из двух АО, которые обозначим, как и в методе ВС, буквами AиB. Тогда любая МО должна выражаться линейной комбинацией типа:
= CAA+CBB
Учет симметрии молекулы приводит к условию: |CA|2= |CB|2, которое выполняется в двух случаях: приCA= +CBиCA= –CB. Следовательно, можно построить всего две МО — одну четную (G) и одну нечетную(U):
G = Cg (A + B) и U = Cu (A – B)
Нормировочные множители можно найти стандартным путем:
Cg= 1 / (2 + 2s)1/2иCu= 1 / (2 – 2s)1/2
Дополнив полученные МО спиновыми множителями или, получим четыре варианта молекулярныхспин-орбиталей(МСО):G,G,UиU.
При сближении атомов водорода их электроны, движущиеся по атомным типам АиВ, вынуждены перейти к молекулярным типам движения, в качестве которых и выступают найденные четыре МСО. Поскольку электронов всего два, заселены будут только две из четырех МСО. Следовательно, существует несколько вариантов конечного состояния молекулы, а именно — шесть.
|
Номер состояния |
1 |
2 |
3 |
4 |
5 |
6 |
|
состояние электрона № 1 |
G |
G |
G |
G |
G |
U |
|
состояние электрона № 2 |
G |
U |
U |
U |
U |
U |
Для каждого варианта можно построить глобальную волновую функцию в виде определителя Слэтера. Например, для первого варианта волновая функция будет иметь вид (без учета нормировочного множителя):

Запишем вид и остальных функций, придерживаясь стандартного соглашения (первый сомножитель относится к первому электрону, второй — к второму).
|
1 |
2 |
Глобальная волновая функция |
|
G |
G |
Ф1 = G G – G G = [GG]( – ) |
|
G |
U |
Ф2 = G U – U G = [GU – UG]() |
|
G |
U |
Ф3 = G U – U G |
|
G |
U |
Ф4 = G U – U G |
|
G |
U |
Ф5 = G U – U G = [GU – UG]( ) |
|
U |
U |
Ф6 = U U – U U = [UU]( – ) |
Все эти волновые функции обладают нужной для выполнения принципа Паули перестановочной антисимметричностью. Глобальные волновые функции, однако, должны кроме этого, обладать и подходящей пространственной симметрией, например, быть либо четными, либо нечетными. Для проверки этой характеристики, необходимо отделить пространственную часть от спиновой и подействовать на пространственную часть оператором инверсии.
Из таблицы видно, что функции Ф3иФ4не разделены на пространственный и спиновой сомножители. Поэтому для них невозможно определить тип симметрии. Обойти эту трудность можно посредством известного приема — симметризации — заменить неправильные функцииФ3иФ4на их сумму и разность, обладающие правильной симметрией:
Ф'3=Ф3+Ф4 = [GU–UG](+)
Ф'4=Ф3+Ф4 = [GU+UG](–)
Теперь можно установить пространственную симметрию глобальных волновых функций:
|
Ф |
Пространственный множитель |
Тип симметрии |
Спиновой множитель |
S |
MS |
|
Ф1 |
GG |
четный (g) |
– |
0 |
0 |
|
Ф2 |
GU – UG |
нечетный (u) |
|
1 |
+1 |
|
Ф'3 |
GU – UG |
нечетный (u) |
+ |
1 |
0 |
|
Ф'4 |
GU + UG |
нечетный (u) |
– |
0 |
0 |
|
Ф5 |
GU – UG |
нечетный (u) |
|
1 |
–1 |
|
Ф6 |
UU |
четный (g) |
– |
0 |
0 |
Видно, что волновые функции Ф2,Ф'3иФ5образуют триплет: их пространственные множители одинаковы, а спиновые состояния отличаются ориентацией вектора глобального спина молекулы.
Фu= (1/2)0,5(GU–UG)[C1() +C2(+) +C3()]