

4.9. Гибридизация с участием d-ао.
В образовании ГАО наряду с s- и p-АО могут участвовать d-АО. Полинг показал, что при составлении линейных комбинаций из одной s-, двух р- и одной d-АО получаются четыре так называемые "квадратные" ГАО (sp2d-гибридизация), которые эквивалентны. Они находятся в одной плоскости и направлены к углам квадрата.
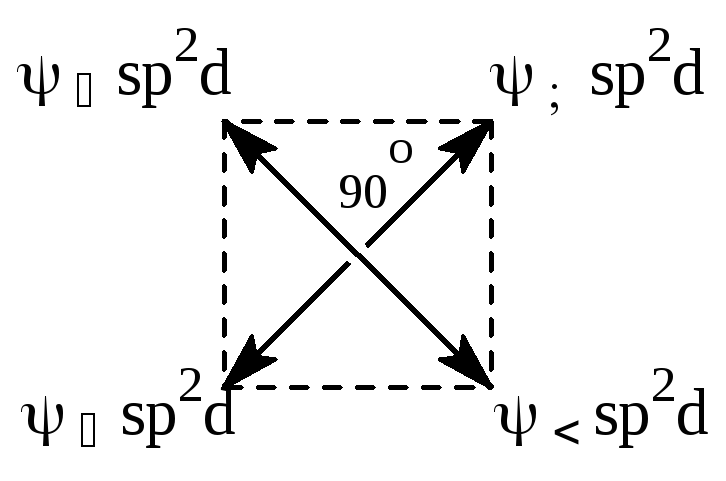
Рис.4.14 Схематическое изображение sp2d-ГАО.
Кроме dsp2-гибридизации иногда наблюдается d2sp3-гибридизация, приводящая к 6 эквивалентным ГАО. Схематически d2sp3-ГАО можно изобразить следующим образом:
Рис.4.15 Схематическое изображение вида d2sp3-ГАО.
Шесть ГАО вида d2sp3 направлены к вершинам правильного октаэдра.
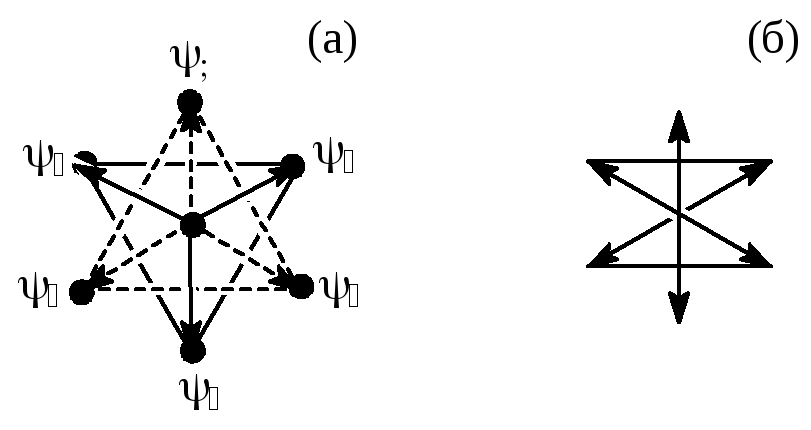
Рис. 4.16 Схематическое расположение в пространстве 6 ГАО d2sp3-типа. (а) - вид "сверху", (б) – сбоку; (в) - снизу.
Возможны и другие типы гибридизации с участием d-АО.
Они приведены в таблице 4.3.
Таблица 4.3 Возможные типы гибридизации с использованием s-, p- и d-АО.
|
Геометрическое расположение |
Число ГАО |
Обозначение ГАО |
|
линейное нелинейное плоское (тригональное) тригонально-пирамидальное тетраэдрическое квадратное тригонально-бипирамидальное плоское пентагональное октаэдрическое додекаэдрическое |
2 2 3 3 4 4 5
5 6 8 |
sp, dp ds sp2, dp2 p3, d2p sp3, sd3 sp2d, p2d2 sp3d, spd3
p2d3, sp3d2 sp3d4 |
Необходимо заметить, что одним из основных условий, необходимых для гибридизации, является близость энергий, участвующей в ней АО. Таким свойством обладают элементы 2-го и 3-го периодов Периодической системы, а также переходные элементы. Именно гибридизацией объясняется способность переходных элементов к образованию комплексных соединений.
4.10. Полуэмпирические методы расчетов с использованием уравнений Хартри-Фока-Руутаана
Объединим модель независимых электронов, нашедшую свое воплощение в уравнениях Хартри-Фока, с приближением МО ЛКАО. Рассмотрим случай четного числа электронов в системе. Уравнения Хартри-Фока имеют вид:
![]() (4.39)
(4.39)
где
![]() (4.40)
(4.40)
оператор Фока или фокиан,
![]()
одноэлектронный оператор в фокиане,
![]()
кулоновский оператор,
![]()
обменный оператор,
M – число ядер в молекулярной системе,
Zj – заряд ядра j в единицах заряда электрона,
Rji – расстояние между ядром j и электроном i,
e – заряд электрона.
Будем искать наилучшую волновую функцию в виде разложения по АО (т.е. в приближении МО ЛКАО):
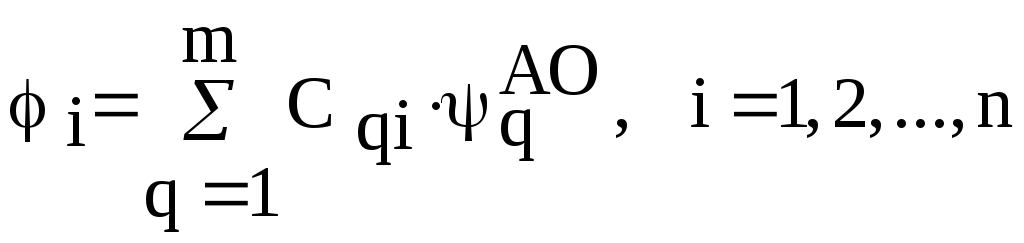 (4.41)
(4.41)
где
m – число атомных орбиталей, по которым
проводится разложение МО,
![]() - АО с номером q,
- АО с номером q,![]() - коэффициент.
- коэффициент.
Подставив разложение 4.41 в 4.39 и 4.40, имеем
![]()
Умножая
полученное соотношение слева на
![]() и приводя интегрирование, приходим к
равенству
и приводя интегрирование, приходим к
равенству
![]() (4.42)
(4.42)
Здесь введены обозначения для матричных элементов
![]()
![]()
Равенство 4.42 можно переписать в матричной форме
![]() (4.43)
(4.43)
Здесь m – число АО в разложении, n – число электронов, F – матрица Фока или фокиан (mn); Сi матрица-столбец коэффициентов для i-го электрона (1n); i – энергия i-ой МО; S – матрица интегралов перекрывания (mn).
Матричные элементы фокиана можно записать в виде
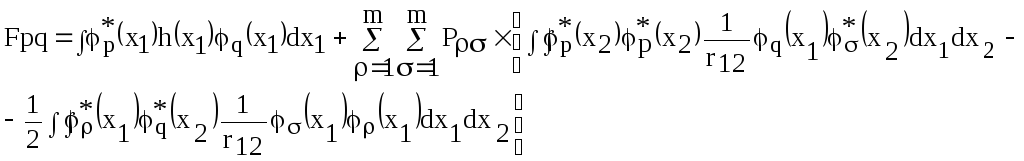
(4.44)
Здесь
![]() (4.45)
(4.45)
матричный элемент порядков связей, aj – заселенность j-ой МО, т.е. количество электронов, которое "ее заселяет".

Здесь был рассмотрен случай замкнутых оболочек. Случай незамкнутых электронных оболочек рассматривается аналогично.
С математической точки зрения сущность метода Руутана состоит в замене решения сложного интегро-дифференциального уравнения Хартри-Фока задачей на собственные вектора и собственные значения матрицы Фокиана. Метод Руутана применим к задачам, не допускающим разделения переменных. С его помощью проведены практические расчеты хартри-фоковских МО самого общего вида. Он произвел революцию в квантовой химии, превратив решение ее задач из искусства, которое "доступно" лишь немногим, в сравнительно легко осуществимую вычислительную процедуру с использованием современных мощных ЭВМ. Кроме того этот метод позволяет получать сколь угодно точные решения уравнения Хартри-Фока и записывать их в форме, удобной для химической интерпретации.
И все же для его практического применения необходимо решить еще несколько проблем.
Одной из них является проблема выбора базисных функций .
Обычно обращают внимание на следующие критерии:
(а) достижение максимальной точности с минимальным числом слагаемых;
(б) удобство вычисления интегралов межэлектронного отталкивания.
В настоящее время качестве функций чаще всего используют следующие АО:
1. Водородоподобные. Преимуществом таких АО является ортонормированность, а также ясность теоретических моделей, которые привели к АО указанного вида. Однако, математические функции для написания таких АО сравнительно сложны, и интегралы межэлектронного отталкивания, вычисленные с использованием водородоподобных АО, считаются весьма долго.
2.
Слейтеровские.
Их радиальные части имеют вид:
![]() ,
гдеn
- главное квантовое число;
- постоянная для рассматриваемого атома
и его электронной оболочки, характеризуемой
квантовым числом n.
Серьезным преимуществом орбиталей
этого типа является их математическая
простота, легкость вычисления интегралов
межэлектронного отталкивания. Недостаток
этих АО заключается в том, что они не
являются ортонормированными.
,
гдеn
- главное квантовое число;
- постоянная для рассматриваемого атома
и его электронной оболочки, характеризуемой
квантовым числом n.
Серьезным преимуществом орбиталей
этого типа является их математическая
простота, легкость вычисления интегралов
межэлектронного отталкивания. Недостаток
этих АО заключается в том, что они не
являются ортонормированными.
3.
Гауссовы.
Их радиальная часть имеет вид
![]() ,
где
- параметр. Преимуществом этих орбиталей
является их простота и легкость вычисления
интегралов межэлектронного отталкивания,
исходя из этих АО. Недостаток метода
заключается в том, что АО плохо
апроксимируются одной простой гауссовой
функцией. Каждую орбиталь приходится
записывать как сумму нескольких гауссовых
функций. Поэтому. хотя интегралы
становятся (по отдельности) более
простыми, их число сильно возрастает.
И, хотя для хорошей аппроксимации АО
необходимо строить линейные комбинации
из 7 гауссовых АО, современные компьютерные
программы такую аппроксимацию используют,
поскольку интеграл межэлектронного
отталкивания, построенный из гауссовых
функций, считается в 106
раз быстрее, чем интеграл, построенный
из слэтеровских АО.
,
где
- параметр. Преимуществом этих орбиталей
является их простота и легкость вычисления
интегралов межэлектронного отталкивания,
исходя из этих АО. Недостаток метода
заключается в том, что АО плохо
апроксимируются одной простой гауссовой
функцией. Каждую орбиталь приходится
записывать как сумму нескольких гауссовых
функций. Поэтому. хотя интегралы
становятся (по отдельности) более
простыми, их число сильно возрастает.
И, хотя для хорошей аппроксимации АО
необходимо строить линейные комбинации
из 7 гауссовых АО, современные компьютерные
программы такую аппроксимацию используют,
поскольку интеграл межэлектронного
отталкивания, построенный из гауссовых
функций, считается в 106
раз быстрее, чем интеграл, построенный
из слэтеровских АО.
Квантово-химические расчеты, в которых используется метод Хартри-Фока-Руутана, часто называют неэмпирическими или ab initio. Широкому их использованию препятствуют следующие принципиальные трудности:
1) Число интегралов межэлектронного отталкивания пропорционально 4 степени от числа АО. С увеличением числа АО возрастают математические сложности.
2) Ошибка при вычислении энергии атомизации молекулы даже при точном решении уравнения Хартри-Фока-Руутана составляет около 1%.
Причина последнего заключена в использовании в теории модели независимых электронов. Считается, что частица движется в усредненном поле всех других частиц, т.е. не учитывается корреляция между положениями электронов. В последние годы эта проблема успешно решается.
Хотя, начиная с середины 90-х годов ХХ века, неэмпирические расчеты стали применяться повсеместно, для получения полуколичественных результатов для систем, содержащих большое число атомов, используются так называемые полуэмпирические методы. Переход к ним осуществляется двумя, часто дополняющими друг друга, способами. Во-первых, вводятся приближения, при использовании которых существенно уменьшается число учитываемых интегралов. Во-вторых, вместо вычисления некоторых интегралов подставляются их численные значения, подобранные таким образом, чтобы полуэмпирическая теория воспроизводила экспериментальные характеристики веществ, выбранных для калибровки параметров.