

5.3.1 Методы триангуляции
Пик рассматривают как треугольник и площадь его рассчитывают как площадь треугольника. Известны три метода триангуляции (triangle – треугольник). Эти методы приближенные, поскольку площадь пика аппроксимируется площадью одного из трех треугольников.
а). В первом методе триангуляции за площадь пика принимается площадь описанного треугольника, образованного касательными, проведенными к сторонам пика в точках перегиба, и отрезком, отсекаемым касательными на основании пика (рис. 30, а).
Площадь описанного треугольника рассчитывается обычно по формуле:
S
= ½ ⋅
h'
⋅
b0
(65)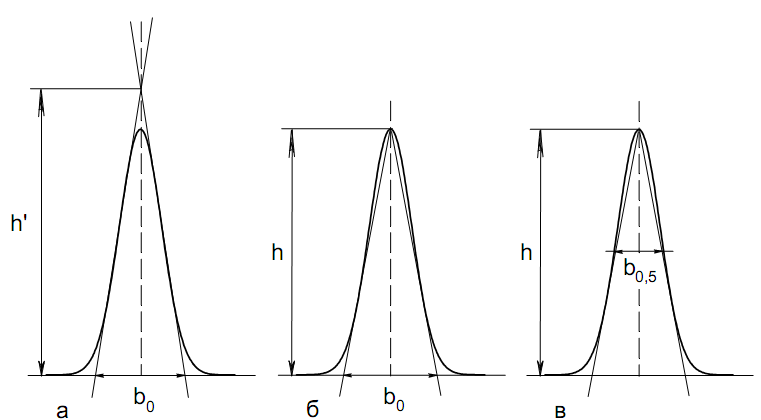
где h' – высота описанного треугольника; b0 – ширина пика на нулевой линии.
Этот метод дает около 97% (96,8%) от площади гауссова пика. Недостатки этого метода состоят в необходимости дополнительных геометрических построений (проведение касательных), которые не всегда могут быть выполнены легко и с достаточной точностью. Особенно это касается узких пиков. При этом точка пересечения касательных может оказаться за пределами диаграммной ленты.
б). Во втором методе триангуляции (рис. 30, б) предложено экстраполировать «прямолинейные» участки ветвей пика только вниз до пересечения с основанием и площадь рассчитывать по формуле:
S = ½ ⋅ h ⋅ b0 (66)
Этим методом находят около 80% (79,8%) от площади гауссового пика. Здесь частично преодолеваются недостатки первого метода.
в). На практике наибольшее распространение получил третий метод триангуляции (рис. 30, в). Метод произведения высоты на ширину пика на половине его высоты. В этом случае геометрические построения упрощаются. Процесс измерения состоит из четырех операций, а именно:
– проведение основания под пиком (интерполирование нулевой линии между началом и концом пика);
– измерение высоты пика;
– нахождение середины высоты;
– измерение ширины пика на половине высоты.
Площадь рассчитывается по формуле:
S = h ⋅ b0,5 (67)
Этим методом находят около 94% (93,9%) от площади гауссова пика. Точность измерения площади в методе «h ⋅ b» зависит от формы и абсолютных размеров пика. Форму пика принято характеризовать отношением «h / b0,5» . Ошибка минимальна при h / b0,5 равна 5−6%. С повышением абсолютных размеров пика точность измерения площади пика возрастает.
Истинная же площадь гауссова пика может быть найдена по формуле:
Sист = h⋅b0,368 (68)
5.3.4 Определение площадей не полностью разделенных пиков
Рассмотрим случай наложения двух гауссовых пиков, когда огибающая имеет минимум (рис. 32). При наложении двух пиков происходит искажение измеряемых параметров пика (высоты и ширины). При этом степень искажения параметров зависит от полноты разделения и соотношения высот соседних пиков. Известны различные приемы расчета хроматограмм с не полностью разделенными пиками.
Метод Бартлета и Смита (корректировки высоты)
Площадь
неразделенных пиков рассчитывается по
обычной формуле для гауссовых пиков,
например по формуле (73). Однако в формулу
подставляют значения истинных параметров
пиков, а не измеренных линейкой на
хроматограмме. Корректировка высоты
пиков, учитывающая их взаимное влияние.
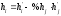
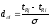
Метод опускания перпендикуляра
При взаимном наложении двух пиков площадь под огибающей кривой равна сумме площадей под индивидуальными пиками (рис. 26). В этом методе границей двух не полностью разделенных пиков является перпендикуляр, опущенный из минимума огибающей кривой на основание пиков. За площадь первого пика принимается площадь, лежащая слева от перпендикуляра, а за площадь второго пика принимается площадь, лежащая справа. Этот метод приближенный, поскольку перпендикуляр является истинной границей только в случае неразделенных пиков одинаковой высоты и ширины. Для того чтобы исключить систематические ошибки, измеренные площади умножают на значения поправочных коэффициентов.
В теории хроматографического анализа следует сделать два допущения:
Во-первых, предполагается идентичность состава пробы, введенной в хроматограф, и смеси анализируемых веществ. Это можно выразить равенством массовых долей определяемого компонента.
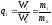 (78)
(78)
где Wi и mi – масса определяемого компонента в отобранной пробе анализируемых веществ и дозируемой пробе соответственно; Wn и mn – навеска анализируемой пробы и хроматографирумой пробы, соответственно.
Во-вторых, предполагается линейная зависимость между параметрами пика и содержанием компонента в пробе:
mi = kai ⋅ Si , (79)
где kai – коэффициент пропорциональности (абсолютный калибровочный коэффициент); Si – площадь пика.
В линейном динамическом диапазоне детектора ki величина постоянная и от величины пробы не зависит. Объединяя предыдущие два уравнения, получаем:
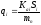 (80)
(80)
Итак, для определения состава анализируемой смеси необходимо учитывать различную чувствительность детектора к разным веществам, измерить количественный параметр пика и учесть массу пробы. Различные методы расчета состава смесей по хроматограммам, как будет видно из дальнейшего изложения, отличаются способом учета величины пробы.
Известны
четыре основных метода расчета состава
смеси по хроматограммам: метод абсолютной
калибровки, метод внутренней нормализации,
метод внутреннего стандарта и метод
стандартной добавки.
Метод абсолютной калибровки основан на том, что для, количественного определения содержания компонентов в анализируемой пробе, необходимо для каждого определяемого в пробе индивидуального вещества построить калибровочную кривую зависимости площади или высоты хроматографического пика от содержания этого вещества в анализируемой пробе. Необходимость такой трудоемкой работы обусловлена тем, что чувствительность детектора хроматографа к различным веществам даже близкой химической структуры, как правило, неодинакова.
Построение калибровочных кривых производят по результатам хроматографирования в одинаковых условиях известных количеств определяемого вещества. Зависимость площади Si или высоты хроматографического пика от содержания qi вещества в анализируемой пробе при достаточно строгом воспроизведении условий хроматографирования является линейной и угловой коэффициент (тангенс угла наклона) калибровочной прямой i-го компонента называется калибровочным коэффициентом Ki, г/см2:
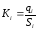 (1.59)
(1.59)
Процентное содержание i-го компонента Xi в пробе составит
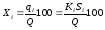 (1.60)
(1.60)
где Q масса анализируемой пробы, г.
Метод абсолютной калибровки достаточно прост, но точность его в значительной мере зависит от постоянства режима и тщательности приготовления и анализа эталонов или их смесей.
Недостаток метода состоит в том, что приходится строить калибровочные графики для каждого индивидуального вещества, на что тратится много времени и расходуются эталонные вещества. Поэтому его применяют, главным образом, при определении одного или нескольких компонентов смеси, например, микропримесей.
Метод внутреннего стандарта (метод метки) основан на сравнении высот или площадей пиков известного вещества-метки и определяемых компонентов. В качестве внутреннего стандарта (метки) стараются подобрать такое вещество, которое бы не реагировало с компонентами смеси, не очень сильно сорбировалось и появлялось на хроматограмме отдельно от других компонентов. Кроме того, его не должно быть в составе исследуемой смеси.
К анализируемой пробе добавляют точно известное количество вещества-метки. Количество метки подбирают такое, чтобы площадь его пика была соизмерима с площадью пиков компонентов смеси, подлежащей количественному определению. Для калибровки следует провести хроматографический анализ ряда смесей вещества-метки с каждым из отдельных определяемых компонентов при различных соотношениях обоих веществ в смеси. По оси абсцисс откладывают значения отношения массы метки qm к массе анализируемого вещества qi (qm /qi) в искусственных смесях, а по оси ординат соответствующее им соотношение площадей пиков Sm/Si.
В узкой области соотношений qm /qi график прямолинеен. Угловой коэффициент такого графика Кi представляет собой калибровочную константу для данного определяемого компонента. Он вычисляется из уравнения
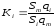 . (1.61)
. (1.61)
Отсюда
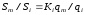 .
.
Калибровочная константа практически не зависит от количества добавляемой в колонку искусственной смеси и мало изменяется при изменении скорости потока подвижной фазы и температуры колонки. Относительное содержание вещества-метки Im в смеси рассчитывается по следующей формуле:
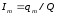 . (1.62)
. (1.62)
Относительное содержание определяемого компонента Ii
 , (1.63)
, (1.63)
где
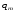
масса вещества-метки в пробе;
масса вещества-метки в пробе;
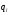
масса определяемого вещества в пробе;
Q
весовое количество пробы смеси, введенное
в колонку.
масса определяемого вещества в пробе;
Q
весовое количество пробы смеси, введенное
в колонку.
Разделив Im на Ii получаем
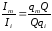 ,. (1.64)
,. (1.64)
откуда
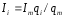 .
.
Процентное содержание определяемого вещества вычисляется из уравнения
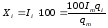 , (1.65)
, (1.65)
а
так как
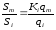 ,
то
,
то
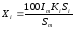 . (1.66)
. (1.66)
Таким образом, чтобы провести анализ и количественный расчет методом метки, нужно:
1) составить
ряд искусственных смесей определяемого
вещества с веществом-меткой, снять ряд
хроматограмм при режиме анализа смеси
и измерить соотношение площадей
соответствующих пиков; на основании
полученных данных построить графики в
координатах
 ;
рассчитать калибровочную константуKi;
;
рассчитать калибровочную константуKi;
2) определить приблизительное весовое количество анализируемой смеси, которое нужно вводить в колонку, чтобы получить достаточно хорошо выраженный пик определяемого компонента; подобрать такое количество вещества-метки, при котором на хроматограмме появился бы пик метки, соизмеримый с пиком определяемого компонента; приготовить стандартную смесь метки и компонентов; вычислить Im по следующей формуле:
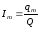 . (1.67)
. (1.67)
3) в хроматографическую колонку ввести пробу анализируемой смеси с известным содержанием метки и снять хроматограмму; измерить площадь пика определяемого компонента Si и пика вещества метки Sm; зная калибровочную константу Ki, рассчитать процентное содержание компонентов по формуле
 (1.68)
(1.68)
Если необходимо определить несколько компонентов, то для каждого из них находят калибровочную константу описанным способом.
Метод метки удобен тем, что на результаты анализа колебания параметров хроматографического опыта сказываются слабо. Но если дозировка объемов анализируемых образцов проводится достаточно точно, этот метод никаких преимуществ перед методом абсолютной калибровки не имеет. Кроме того, использование метода метки ограничивается преимущественно определением лишь небольшого числа компонентов, содержание которых в не очень сложных по составу смесях не превышает примерно 10%.
Метод простой нормировкиможно использовать при количественном обсчете хроматограмм в том случае, когда есть уверенность в том, что все вещества анализируемой пробы, взятые в равном количестве, дают на хроматограмме пики, имеющие равные площади, т. е. что чувствительность детектора хроматографа ко всем компонентам анализируемой пробы одинакова. Это условие достаточно строго выполняется, если вещества сходны по химическому строению. Площади пиков рассчитывают, умножая высоту пика на ширину, измеренную на его полувысоте. Процентное содержание данного компонента в анализируемой смеси находят по формуле:
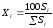 (1.69)
(1.69)
где Xi процентное содержание i-ого компонента; Si площадь пика определяемого компонента; Si сумма площадей пиков всех компонентов.
Метод простой нормировки не дает точных результатов в случае различной чувствительности детектора по отношению к различным компонентам смеси. В этом случае следует применять метод внутренней нормировки с калибровочными коэффициентами.
Этот метод учитывает различия чувствительности детектора по отношению к компонентам анализируемой смеси. Он более точен и надежен, чем метод простой нормировки. Количественный обсчет хроматограмм ведут по формуле
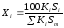 , (1.70)
, (1.70)
где Кi калибровочный коэффициент, мг/см2.
Если отдельные компоненты разделяются не полностью, то пользуются следующей формулой:
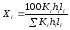 , (1.71)
, (1.71)
где hi высота пика i-ого компонента; li расстояние удерживания этого компонента (по диаграммной ленте).
Физический смысл калибровочного коэффициента Ki количество вещества, соответствующее единице площади пика.
Практически калибровочный коэффициент рассчитывают следующим образом. Приготовив искусственную смесь определенного состава, снимают хроматограмму. Разделив массу каждого компонента в пробе этой смеси на площадь соответствующего пика компонента, получают калибровочный коэффициент Ki (мг/см2).
Для определения Ki часто используют так называемые поправочные коэффициенты Ki’, т.е. калибровочные коэффициенты Ki хроматографируемых веществ, к калибровочному коэффициенту Kст. вещества, выбранного в качестве стандарта:
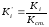 . (1.72)
. (1.72)
K’стявляется безразмерной величиной.
Применение метода внутренней нормировки при использовании большинства детекторов часто не требует специальной калибровки, так как калибровочные коэффициенты для многих веществ даны в литературе. Для быстрого определения приведенной площади пика КiSi можно воспользоваться имеющимися в литературе номограммами.
Достоинство метода внутренней нормировки состоит в том, что искажения, имеющиеся в одинаковой степени у всех пиков, в конечном счете не влияют на точность результатов. Ошибки этого метода могут быть связаны с постепенным изменением режима в процессе анализа.