

Параметр а
Влияние изменения условий хроматографических разделений на параметр А (площадь пика) сводятся к следующему:
флуктуации температуры не искажают площадь пика при работе с детекторами обоих типов;
флуктуации скорости потока газа-носителя при использовании концентрационных детекторов могут искажать как абсолютные, так и относительные значения параметра А;
перегрузка колонки пробой, если она не ухудшает разделения до недопустимых пределов, практически не влияет на площадь пика.
Площадь пика для детекторов, используемых в количественной хроматографии, является универсальным параметром, применимым во всех случаях, когда отсутствуют необратимые процессы в колонке и когда детектор работает в области нормальных рабочих условий.
Применение потоковых детекторов при использовании параметра А предпочтительнее.
Имеются сведения о количественной характеристике устойчивости различных параметров пика. Так, изменение относительной высоты пика составляет 0.30.9 % при изменении температуры на 1 К. Точность 2 % при анализе методом абсолютной калибровки по высотам пиков может быть достигнута при стабилизации температуры с точностью до 0.1 К и скорости газа-носителя до 1 мл/мин. Трудно получить результаты с относительной погрешностью менее 2.8 3.2 % при концентрации компонента в пробе 12 %, если в качестве параметра пика используется его высота.
Для детектора по теплопроводности в сочетании с измерением площади пика получены следующие значения допустимых погрешностей задания параметров (при условии получения погрешности результата не более 1 %):
Т а б л и ц а 12
Величины допустимых погрешностей задания параметров разделения
|
давление на входе в колонку |
|
|
давление на выходе из колонки |
|
|
ввод пробы |
|
|
температура |
|
|
ток моста детектора |
|
|
погрешность измерения площади пика |
|
 0.10
%
0.10
% 0.22
%
0.22
% 0.41
%
0.41
% 0.23
%
0.23
% 0.14
%
0.14
% 0.41
%
0.41
%
Эти данные получены с учетом влияния каждого фактора на конечный результат анализа. При этом вклад в погрешность результата всех шести источников частных погрешностей предполагался одинаковым.
Если, например, измерять не площадь пика, а высоту пика, то допустимая погрешность изменения температуры оказывается в 2 раза меньшей.
Большой вклад в погрешность вносит давление на выходе из колонки. Поскольку эта величина определяется изменением атмосферного давления, ее колебания могут быть достаточно велики (до 3 %). Между тем, в практической хроматографии это часто не учитывается. В результате возникают существенные погрешности при использовании для расчетов абсолютных значений параметров. По этой причине детектор ионизации в пламени считается более пригодным для количественных измерений, чем детектор по теплопроводности.
Для получения погрешности менее 1 % при определении времени удерживания требуются не столь жесткие условия по стабильности аппаратуры: давление на входе в колонку можно поддерживать с точностью 0.4 %, температуру с точностью 0.5 К и выполнять измерения величины площади с точностью 0.5 %.
5.2
ИЗМЕРЕНИЕ КОЛИЧЕСТВЕННЫХ ПАРАМЕТРОВ
ХОРОШО РАЗДЕЛЕННЫХ ПИКОВ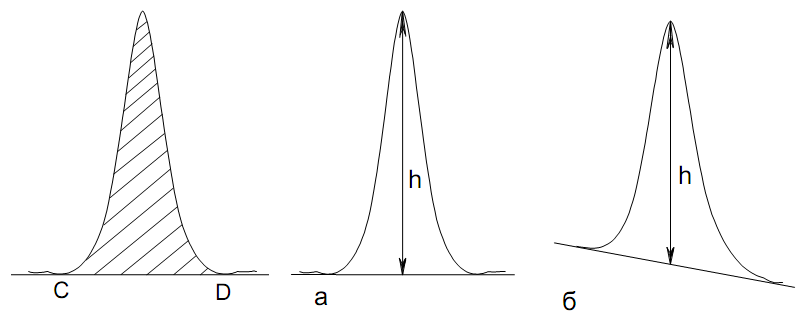
Количественный газохроматографический анализ основан на зависимости между площадью S (или высотой h) пика и количеством определяемого компонента в пробе. Под площадью пика понимается площадь, ограниченная контуром пика и его основанием — СD отрезком, соединяющем начало и конец пика (рис. 24). Площадь пика измеряется в мм2, А⋅с или В⋅с. Высота пика (h) — расстояние от вершины пика до его основания, измеренное в направлении параллельной оси сигналов детектора (рис. 25), h – измеряется в мм: а – нулевая параллельная оси времени, т.е. дрейф отсутствует (рис.25, а); б – измерение высоты при дрейфе нулевой (рис. 25, б).
Площадь и высота пика могут измеряться как вручную, так и автоматически. При ручных способах измерения площади пика используют вспомогательный параметр - ширина пика (рис. 26).
Ширина
пика - проекция отрезка прямой, параллельной
основанию пика, ограниченного точками
пересечения с ветвями пика, на ось
времени. Ширина пика может измеряться
на разных уровнях (сечениях) высоты
пика. Ширина пика измеряется в мм; сек.
Наиболее просто вручную измеряются и
рассчитываются параметры так называемых
гауссовых пиков (рис. 27). Контур этих
пиков описывается уравнением: где х, у
– координаты точки контура пика; h и S –
высота и площадь пика, отвечающая
максимальной концентрации компонента
в зоне; σ – стандартное отклонение,
которое отвечает ширине пика на высоте
0,882 h.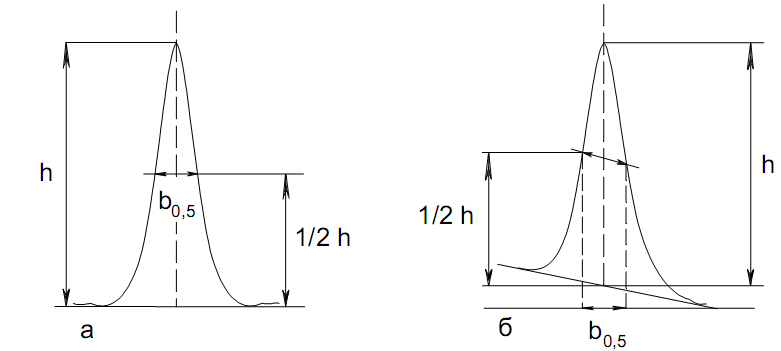
Стандартное отклонение может быть определено также из соотношений, справедливых для гауссовских пиков:
2σ = b0,607 2,36σ = b0,500 3σ = b0,324 4σ = b0,134
Расчет площади пика как площади, ограниченной гауссовой кривой (для симметричных пиков), проводят по формуле, полученной интегрированием гауссовой функции распределения ошибок:
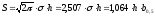 (62)
(62)
где S – площадь пика; h – его высота; σ – стандартное отклонение, равное ширине пика на высоте b0,882.
Точность
измерения площади пика этим методом
определяется точностью измерения
отрезков на хроматограмме (h и b0,882),
а, поскольку, высота пика обычно много
больше ширины пика, то точность измерения
площади пика в значительной степени
определяется точностью измерения
ширины пика. Поэтому при измерении
ширины узких пиков (меньше 10 мм) желательно
пользоваться измерительной лупой и
учитывать толщину линии.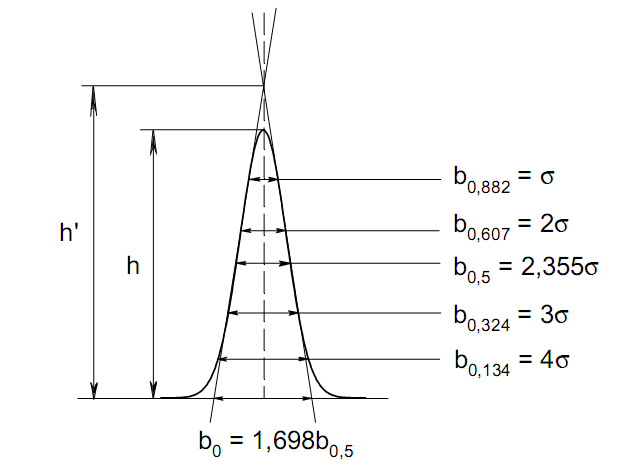
Обработка
хроматограмм с асимметрическими пиками,
как правило, проводится с меньшей
точностью. Асимметричность пика
анализируемого соединения может быть
вызвана двумя причинами: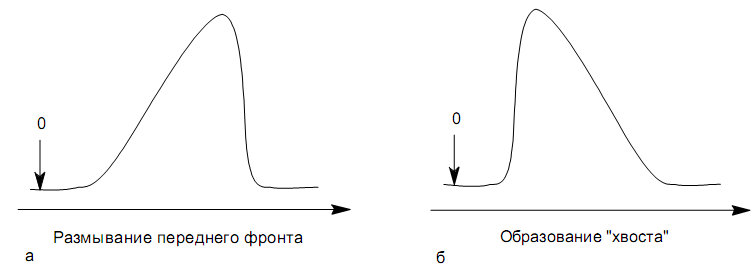
1. Перегрузкой колонки анализируемым веществом.
2. Наличием остаточной адсорбционной активности твердого носителя, используемого для приготовления сорбента.
Форма пиков при этом различна. Перегрузка колонки приводит к образованию размытого фронта (рис. 28, а). Остаточная адсорбционная активность приводит к образованию хвоста (рис. 28, б).
Устранение асимметричности первого рода достигается при уменьшении дозы. В то время как устранение асимметричности второго рода более трудная задача. Для численного выражения асимметричности предложено использовать коэффициент асимметричности (рис. 29):
Kas=SA/SB=c/d=a/b (63)
где
SA и SB – площади между осевой линией пика
и задним его фронтом, и передним его
фронтом соответственно; а и b –
расстояния между осевой линией пика и
задним его фронтом и передним его фронтом
на полувысоте соответственно; c и d – то
же самое на 0,1 высоты пика.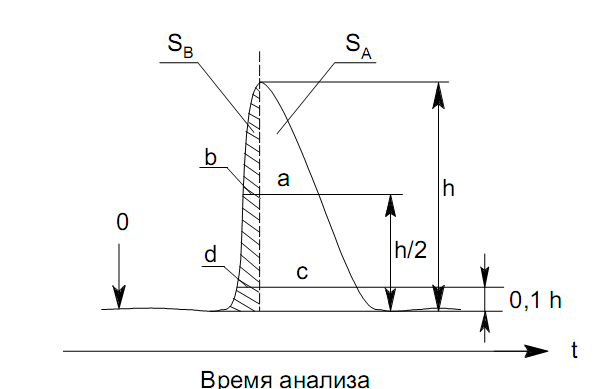
Для симметричного пика Каs = 1. В случае остаточной адсорбции Каs > 1, при «перегрузочных» пиках Каs < 1. Считается допустимым работать на колонке, имеющей Кas для всех компонентов анализируемой смеси в пределах 0,7 – 1,5. Искажение формы пика при адсорбции («хвостование») зависит и от природы анализируемых соединений. Менее всего адсорбируются насыщенные углеводороды, наиболее сильно - высокополярные соединения. Аналитическая колонка может оказаться пригодной для анализа соединений одного класса и совершенно не пригодной для соединений другого класса. Ее следует охарактеризовать и по этому признаку.