

В присутствии (а) и в отсутствие (б) Br в составе ведущего электролита.
Это является сигналом к тому, чтобы заново приготовить свежий раствор ДЭА (быстрее всех поглощает СО2) или полностью заменить компоненты ведущего электролита на свежеприготовленные. Количественное определение НСО3 невозможно из-за его неконтролируемого содержания в используемых растворах.
1.9. Качественный хроматографический анализ
Задача качественного хроматографического анализа состоит в том, чтобы установить принадлежность полученных на хроматограмме пиков конкретным химическим соединениям. Функция хроматографической колонки в хроматографии сводится лишь к разделению анализируемого вещества на индивидуальные компоненты. Определение их качественного состава проводится за пределами колонки и может быть осуществлено или по характеристикам удерживания каждого из компонентов в данной колонке при конкретных условиях разделения, или с использованием других аналитических приемов. В первом случае на выходе из хроматографической колонки компоненты анализируемой пробы проходят через детектор и фиксируются в виде хроматограммы. Во втором случае компоненты анализируемой пробы на выходе из колонки направляются в какой-либо анализатор, где и анализируются одним из химических или физических методов, их сочетанием, или сочетанием хроматографических, химических и физических методов.
Для детальной идентификации компонентов сложной пробы часто недостаточно провести ее разделение на одной хроматографической колонке, а приходится прибегать к сложной схеме анализа, включающей целый комплекс методов. Часто применяют многоступенчатые схемы анализа, которые позволяют не только идентифицировать компоненты пробы, но и ускорить и удешевить анализ.
В зависимости от состава анализируемой смеси, а также имеющейся аппаратуры и эталонных веществ можно использовать различные методы идентификации.
I. Методы идентификации на одной колонке.
I.1. Применение индивидуальных эталонных веществ или их смесей
Один из вариантов этого метода состоит в последовательном разделении в одинаковых условиях анализируемой смеси и эталонной смеси. Метод можно использовать, если имеются веские основания для предположения о составе эталонной смеси, которые могут быть получены, как правило, кропотливыми предварительными исследованиями. Причиной для идентификации компонентов смеси в этом случае служит равенство времени удерживания пиков соответствующих компонентов анализируемой и эталонной смесей. Если расход газа-носителя при проведении анализов этих смесей неодинаков, то вместо времени удерживания для идентификации пиков используют значения исправленного времени удерживания или исправленных удерживаемых объемов.
Второй вариант этого метода заключается в том, что в анализируемую пробу вводят эталонный компонент, наличие которого в этой смеси предполагается. Увеличение высоты соответствующего пика без его существенного расширения (размытия) по сравнению с высотой этого пика на хроматограмме, полученной до введения эталона, может служить указанием на присутствие искомого соединения в анализируемой смеси.
Данный метод прост методически, но имеет следующие недостатки:
необходимо иметь эталонные вещества;
все пики, полученные при разделении на используемой колонке, должны гарантированно соответствовать индивидуальным веществам.
И даже при соблюдении этих условий нет абсолютных гарантий однозначного проведения идентификации, так как практически всегда имеются по меньшей мере два вещества, удерживаемые объемы которых на применяемой колонке близки, и такими веществами вполне могут оказаться любой компонент смеси и вещество, использованное в качестве эталона, которые между собой не тождественны. Для окончательной идентификации компонентов пробы необходимо подтверждение их химической индивидуальности химическими или физическими методами.
I.2. Использование табличных данных о характеристиках удерживания
В настоящее время опубликовано большое количество таблиц относительных удерживаемых объемов для самых разных веществ. Эти таблицы, а также компьютерные базы данных можно использовать для идентификации компонентов анализируемых проб при отсутствии необходимых эталонных веществ. Анализируемую пробу разделяют на колонке при условиях, указанных в соответствующей таблице, предварительно введя в нее небольшое количество вещества, служащего стандартом. Международной комиссией по номенклатуре на I симпозиуме по хроматографии в Лондоне в 1956 г. рекомендован ряд веществ, которые следует использовать в качестве стандартов: пентан для углеводородов, масляная кислота для жирных кислот, метиловый эфир миристиновой кислоты для эфиров высших жирных кислот. По полученной хроматограмме рассчитывают относительные удерживаемые объемы:
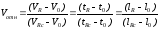 (1.55)
(1.55)
где VR , Vo,VR с. удерживаемые объемы соответственно компонентов пробы, несорбирующейся подвижной фазы и стандарта; tR, to, tR с. время удерживания компонентов пробы, несорбирующейся подвижной фазы и стандарта соответственно; lR, lo, lR с соответствующие отрезки на хроматограмме.
Для идентификации выделенных компонентов анализируемой пробы сравнивают полученные значения относительных удерживаемых объемов Vотн. с табличными данными. Так как внутри одного класса соединений графическая зависимость Vотн. от числа атомов n обычно прямолинейная, то построив калибровочный график зависимости lg Vотн. от n и определив Vотн. выделенных веществ можно достаточно полно их идентифицировать. Для построения калибровочного графика нужно знать Vотн. для 34 членов данного гомологического ряда.
Если в анализируемой пробе содержатся соединения различных классов, то для их идентификации нужно построить несколько аналогичных калибровочных графиков как для полярной, так и для неполярной неподвижной жидкой фазы. Дальнейшая идентификация осуществляется путем повторного выделения идентифицируемого вещества и исследования его различными физико-химическими методами.
Недостатки этого метода идентификации следующие:
необходимость при анализе пробы точно соблюдать условия разделения, использованные при получении опубликованных данных;
наличие дефицитных стандартов для веществ различных гомологических рядов.
Некоторыми преимуществами по сравнению с описанным выше методом при идентификации компонентов анализируемой пробы обладает метод Ковача.
Суть этого метода заключается в использовании линейной зависимости между логарифмами объемов удерживания и числом углеродных атомов нормальных парафинов, выраженным индексами удерживания I.
При проведении качественного хроматографического анализа с использованием метода Ковача в одинаковых условиях измеряют удерживаемые объемы трех веществ идентифицируемого и двух нормальных алканов, различающихся по числу углеродных атомов на единицу. При этом н-алканы выбирают таким образом, чтобы удерживаемый объем идентифицируемого компонента имел промежуточное значение между соответствующими характеристиками н-алканов. Затем из графика (рис. 1.27) или по интерполяционной формуле
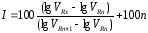 (1.56)
(1.56)
находят индекс удерживания идентифицируемого вещества и по справочным таблицам по величине индекса удерживания определяют, какому веществу принадлежит это значение.
Идентификация по индексам удерживания по сравнению с другими методами идентификации имеет следующие преимущества:
– в качестве стандарта используется не случайное вещество, а гомологический ряд нормальных углеводородов, благодаря чему точность и воспроизводимость определения индексов очень высокие. Кроме того, нормальные углеводороды наиболее доступны в качестве стандартных веществ;
– значения
индексов удерживания намного меньше
зависят от температуры колонки, чем
относительные удерживаемые объемы, что
расширяет диапазон температур колонки,
позволяющий проводить идентификацию;
– при наличии литературных данных по индексам удерживания Ковача можно проводить качественный анализ без применения индивидуальных веществ.
Однако результаты идентификации, полученные и методом Ковача, должны быть проверены другими независимыми методами.
I.3. Использование графических или аналитических зависимостей между относительными удерживаемыми объемами и другими физико-химическими свойствами веществ.
Если между логарифмами относительных удерживаемых объемов веществ одного гомологического ряда и другими физико-химическими характеристиками этих веществ, например температурами кипения, молекулярной рефракцией, молярным коэффициентом экстинкции и др., существует линейная зависимость, то для идентификации компонентов анализируемых веществ могут быть использованы соответствующие графические зависимости.
Например, на колонках с неполярным адсорбентом в последовательности, соответствующей их температуре кипения, выделяются вещества, относящиеся даже к разным классам. На колонках с полярной фазой линейность зависимости логарифма удерживаемых объемов от температуры кипения наблюдается для веществ одного гомологического ряда.
Так как относительный удерживаемый объем является функцией температуры хроматографической колонки, анализ следует проводить именно при тех условиях, для которых построен соответствующий график, иначе идентификация может оказаться неверной, потому что при изменении температуры может измениться даже порядок элюирования. Если рабочая температура отличается от требуемой величины, необходимо привести удерживаемый объем к соответствующей температуре при помощи зависимости lg Vотн. от 1/Т.
Для идентификации хроматографических пиков можно воспользоваться уже опубликованными графиками или построить необходимые кривые на основании результатов разделения калибровочных смесей.
II. Методы идентификации компонентов анализируемых веществ на нескольких колонках
II.1. анализ на параллельных колонках с сорбентами различной полярности
Для
однозначной идентификации компонентов
сложных смесей разработан метод,
основанный на установлении удерживаемых
объемов определяемых веществ на двух
сорбентах различной полярности.
Модификаций этого метода много. Например,
чтобы определить принадлежность
идентифицируе-мого вещества к тому или
иному гомологическому ряду, можно
воспользоваться графиком зависимости
удерживаемых объемов веществ на колонке
с полярной неподвижной фазой от
удерживаемых объемов этих веществ на
такой же колонке с неполярной фазой.
Такой график предварительно строится
для веществ различных классов. При
одинаковом масштабе удерживаемых
объемов на обеих координатных осях для
различных гомологических рядов соединений
получают ряд прямых, проходящих через
начало координат.
Наклон прямых является характеристикой различных гомологических рядов (функциональных групп) (рис.1.28). Идентификация этим методом проводится следующим образом. Сначала устанавливают принадлежность компонента анализируемой смеси к тому или иному гомологическому ряду. Для этого сопоставляют логарифмы или сами удерживаемые объемы идентифицируемого компонента, измеренные на колонках с полярной и неполярной неподвижными фазами. Совпадение характеристик удерживания, которые измеренны по хроматограммам, полученным на обеих колонках, с точкой на той или иной прямой графика указывает на принадлежность компонента гомологическому ряду, соответствующему этой прямой. Положение же точки на этой прямой дает ответ на вопрос, какое это вещество.
Применяется
метод идентификации компонентов пробы,
в котором использована прямолинейная
зависимость между логарифмами объемов
удерживания веществ и безразмерным
параметром Z,
представляющим собой отношение
температуры кипения идентифицируемого
вещества к температуре хроматографической
колонки, при которой проводится
разделение., т. е.
 Сущность
метода сводится к тому, что по индивидуальным
веществам определенного класса соединений
строят калибровочные графики lg
VR
от Z
на различных неподвижных фазах,
отличающихся полярностью. По хроматограммам
анализируемого вещества на этих колонках
определяют удерживаемые объемы
идентифицируемых компонентов. По
калибровочным графикам находят фактор
Z.
Зная температуру колонки Ткол.,
определяют температуру кипения вещества.
Сопоставив полученное значение
температуры кипения со справочными
данными по физико-химическим свойствам
химических соединений, идентифицируют
компонент.
Сущность
метода сводится к тому, что по индивидуальным
веществам определенного класса соединений
строят калибровочные графики lg
VR
от Z
на различных неподвижных фазах,
отличающихся полярностью. По хроматограммам
анализируемого вещества на этих колонках
определяют удерживаемые объемы
идентифицируемых компонентов. По
калибровочным графикам находят фактор
Z.
Зная температуру колонки Ткол.,
определяют температуру кипения вещества.
Сопоставив полученное значение
температуры кипения со справочными
данными по физико-химическим свойствам
химических соединений, идентифицируют
компонент.
II.2. Анализ на последовательно соединенных колонках с сорбентами различной полярности.
При рассмотрении двух или трех хроматограмм одной смеси, полученных на колонках с сорбентами различной полярности, часто очень трудно или даже невозможно установить, какой пик одной хроматограммы соответствует определенному пику другой. Поэтому иногда смесь разделяют на последовательно соединенных колонках с полярными и неполярными сорбентами, а также на составных колонках, состоящих из секций различной длины с указанными сорбентами. При такой схеме анализа можно точно определить характеристики удерживания каждого компонента на неподвижных фазах различной полярности и идентифицировать соответствующие соединения.
III. Сочетание хроматографии с другими методами исследования.
III.1. Определение функциональных групп
При проведении идентификации по удерживаемым объемам на одном сорбенте часто необходимо знать, к какому классу соединений относится идентифицируемый компонент. Это может быть установлено, если выделенный компонент после выхода из хроматографической колонки анализируется с использованием качественных реакций на соответствующие классы соединений или подходящих физико-химических методов. Примеры таких качественных реакций приведены в табл. 1.5.
По наблюдаемому изменению окраски или выпадению осадка судят о функциональной природе идентифицируемого компонента анализируемой пробы. Для окончательной идентификации определяемого вещества часто используют, например, зависимость lg Vотн. от количества углеродных атомов для веществ данного гомологического ряда.
Таблица 1.5
Качественные реакции на некоторые классы соединений, используемые при групповой идентификации
|
Класс соединений |
Реактивы |
Окраска в случае положительной реакции |
|
Спирты |
Дихромат калия + азотная кислота |
Синяя окраска |
|
Альдегиды и кетоны |
2,4-динитрофенил-гидразин |
Желтый осадок |
|
Альдегиды
|
Флороглюцин
|
Оранжево-красный осадок |
|
Кетоны |
Нитропруссид натрия |
Бордово-красный осадок |
|
Амины |
Нитропруссид натрия |
Синяя окраска |
|
Ароматические углеводороды, олефины |
Формальдегид + серная кислота |
Темно-красная окраска
|
|
Галогенпроизводные |
Спиртовой раствор нитрата серебра |
Белый осадок |
III.2. Методики удаления (вычитания).
Идентификацию соединений определенного класса можно осуществить путем использования реагентов, взаимодействующих с этими соединениями с образованием осадка (например, см. табл. 1.2 ), и хроматографического анализа смеси до и после такой обработки. На первой хроматограмме при использовании оптимальнных условий разделения будут содержаться пики всех компонентов, на второй только пики непрореагировавших веществ.
III.3. Использование спектральных методов анализа
Для идентификации компонентов смесей применяется довольно часто их анализ спектральными методами. Наиболее целесообразно для этой цели использовать методы, характеризующиеся высокой специфичностью получаемых спектров, такие как ИК-, ЯМР- и масс-спектроскопия. Эти методы могут применятся для идентификации компонентов анализируемых проб как после их выделения на хроматографической колонке, так и, что представляет наибольший интерес, при непосредственном соединении хроматографа с быстродействующим Фурье-ИК-спектрометром или масс-спектрометром. В некоторых случаях используют и более сложные комбинации, например сочетание газовой или жидкостной хроматографии с детектированием выделенных компонентов ЯМР- и ИК-спектрометром.
Из всех спектральных методов, применяемых для идентификации компонентов в хроматографии, наименее чувствительным является метод ЯМР-спектроскопии. Однако этот метод позволяет получать специфическую информацию для установления структуры анализируемых соединений, например их стереоизомерии.
Наиболее высокая чувствительность анализа может быть достигнута при использовании масс-спектрометрического детектирования компонентов анализируемой пробы, разделенных на хроматографической колонке. Такое сочетание хроматографии с масс-спектрометрией уже нашло широкое применение как в научных исследованиях, так и при контроле качества продукции, получив название хромато-масс-спектрометрия.
III.4. Использование повышенной чувствительности детекторов к некоторым классам соединений
При одновременной работе нескольких детекторов и равенстве количеств вещества, попадающего в каждый детектор, на хроматограммах регистрируются пики различной высоты (и площади) в зависимости от природы анализируемого вещества и принципа действия детектора. Например, пламенно-ионизационный детектор даст для ароматических углеводородов более интенсивный пик, чем катарометр. Детектор электронного захвата очень чувствителен к соединениям, содержащим электроноакцепторные атомы галогенов, свинца и др.
Чувствительность различных детекторов к анализируемым веществам является важным показателем при групповой идентификации компонентов сложных смесей неизвестного состава. При этом используемые детекторы могут быть размещены по отношению к колонке по параллельной или последовательной схеме.
Использование последовательной схемы возможно только при условии, что детектор, соединенный непосредственно с колонкой, является недеструктивным (например, катарометр). Качественная идентификация веществ этим методом основана на определении непосредственно из хроматограмм относительного отклика RD как отношения площадей или высот пиков, зарегистрированных разными детекторами для определяемого компонента, и использовании имеющихся экспериментальных данных по значениям RD для различных анализируемых объектов и детекторов.