

Устройства ввода пробы в хроматограф
Дозатор это устройство для ввода в хроматографическую колонку газовой, жидкой или твердой анализируемой пробы. Дозатор должен удовлетворять следующимтребованиям:
обеспечивать воспроизводимость величины пробы;
его внутренняя поверхность должна быть индифферентна к компонентам анализируемой пробы;
должен быть конструктивно прост, удобен в работе и дешев.
Правильный ввод пробы предполагает обязательное выполнение трех основных требований:
обеспечение минимального размывания пробы в системе ввода пробы;
обеспечение максимальной точности и воспроизводимости дозируемого количества образца;
обеспечение неизменности количественного и качественного состава смеси до и после дозирования.
Первое требование исходит из того, что в упрощенной теории линейной хроматографии идеальная модель исключает какое-либо размывание пробы в системе ввода, поскольку предполагается, что образец в начале хроматографической колонки занимает объем неподвижной фазы, эквивалентный одной теоретической тарелке.
Теоретическая тарелка характеризует такую часть колонки по высоте, на протяжении которой однократно реализуется процесс перехода исследуемого соединения из подвижной фазы в неподвижную и обратно.
На практике конечный объем пробы и конечное время дозирования препятствуют этому. Тем не менее при стремлении к идеальной модели следует вводить минимально возможные по объему пробы за минимально короткий промежуток времени.
Для практической нелинейной хроматографии мгновенный ввод пробы не всегда приводит к оптимальным результатам, поскольку чем выше концентрация компонента на слое сорбента, тем сильнее размывание полосы, когда изотерма адсорбции исследуемого соединения в этой области его концентраций нелинейна.
Таким образом, при мгновенном вводе пробы разбавление образца газом-носителем будет гораздо меньшим, чем при медленном дозировании, и, следовательно, в первом случае в колонку войдет узкая полоса с высокой концентрацией, а во втором – более широкая полоса с меньшей концентрацией.
Оптимальное соотношение этих двух противоположно действующих факторов – ширины полосы и концентрации, обусловленное степенью влияния каждого из них на размывание полосы, определяет время дозирования.
Следует учитывать, что существенное влияние на размывание пробы в системе ввода пробы оказывает конструкция дозатора. В соответствии с этим основными требованиями, предъявляемыми к конструкции дозатора, являются следующие:
минимальный внутренний объем дозатора;
отсутствие непродуваемых газом-носителем полостей во внутреннем объеме дозатора;
хорошо сформированный поток газа-носителя должен быстро переносить весь анализируемый образец непосредственно в колонку.
Второе требование к вводу пробы предполагает дозирование образца с высокой точностью и воспроизводимостью, поскольку хроматография является сравнительным методом анализа. Это требование усугубляется стремлением к вводу минимального количества образца, что на современном уровне составляет примерно 1 мкл газовой пробы и 0.05 мкл жидкой пробы.
Третье требование к вводу пробы предусматривает исключение изменения качественного состава пробы и количественного соотношения анализируемых компонентов в системе ввода, например, за счет разложения при контакте с нагретыми металлическими стенками испарителя, каталитических превращений, полимеризации, селективной сорбции.
В целях устранения этих помех следует:
использовать полностью стеклянные (еще лучше кварцевые) системы ввода пробы;
ввод пробы целесообразно осуществлять непосредственно в хроматографическую колонку;
температура зоны испарения обязательно должна быть выше температуры кипения самого высококипящего компонента.
Следует обязательно учитывать, что при недостаточно высокой температуре в зоне испарения образца может происходить процесс фракционирования пробы. При этом тяжелые компоненты пробы не могут испариться мгновенно, и поступающий в первый момент в колонку пар будет обеднен ими. В особых случаях температура испарителя может программироваться, если стоит задача фракционирования сложной по составу анализируемой смеси уже в испарителе.
Кроме отмеченных общих требований имеются и специфические требования к дозирующим устройствам для каждого из агрегатных состояний проб: газообразных, жидких, твердых.
В зависимости от агрегатного состояния анализируемой пробы используются различные способы их ввода.
Ввод газообразных проб можно осуществить либо с помощью обычного медицинского шприца, либо используя специальные дозирующие устройства.
Использование шприца приводит к существенным ошибкам вводимых объемов пробы ( 10 %) вследствие того, что конец иглы шприца открыт и давление в шприце равно атмосферному, в то время как давление в устройстве для ввода пробы выше атмосферного, и поэтому выше, чем во внутреннем объеме шприца. Следовательно, для ввода пробы необходимо создать поршнем давление большее, чем на входе в хроматограф. Поэтому остаточный объем газа в игле шприца всегда находится при повышенном давлении, его количество отличается от значения при нормальных условиях и не является постоянным.
Специальные дозирующие устройства подразделяются: газовый кран, газовый шток, газовая петля.
При использовании этих дозирующих устройств анализируемая проба становится частью объема газа-носителя и вместе с ним поступает в колонку.
Устройство газового крана приведено на рис. 3.
Сначала анализируемая газообразная смесь, подаваемая из газометра или газопровода под постоянным давлением, заполняет внутренний объем канала 1–2 газового крана. Затем поворотом крана канал с анализируемой газовой смесью помещается в поток газа-носителя и поступает в колонку.
Рис.
3. Схема
устройства газового крана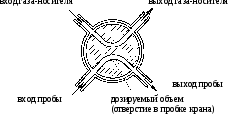
Рис.
4. Схема
устройства газового штока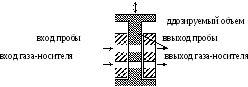
На рис. 4 приведена схема дозирующего устройства с движущимся штоком.
Основным недостатком обеих конструкций является невозможность изменения величины дозирующего объема.
Этого недостатка лишен кран-дозатор со сменными дозирующими газовыми петлями. Схема крана-дозатора приведена на рис. 5.