

Учебник
.pdf
|
|
костном мозге £50%, доля бластов среди |
|
|
|
неэритроидных клеток - более 30% |
|
M7 |
Острый мегакариоцитарный |
Морфологические черты мегакариобластов, |
|
лейкоз |
CD4V, CD6V |
||
|
Дифференциация острых миелоидных лейкозов (М1-М5 и большинства случаев М6) проводится на основании морфологии и цитохимии, которые позволяют охарактеризовать линейную направленность дифференцировки лейкозных клеток (гранулоцитарная, моноцитарная, эритроидная) и
определить степень этой дифференцировки. Морфологическая находка,
высокоспецифичная для острого миелобластного лейкоза, – палочки Ауэра.
Если реакция на миелопероксидазу отрицательна, что характерно для варианта М0, и обнаруживают палочки Ауэра, необходимо выставить диагноз острого лейкоза варианта М1. При вариантах М1 и М2 с t(8;21) часто наблюдают длинные нежные нитеподобные палочки Ауэра; при варианте М3 в цитоплазме можно увидеть пучки этих палочек.
Иммунологические признаки миелоидной дифференцировки включают нелинейные маркёры гемопоэтических предшественников CD34 и HLA-DR,
панмиелоидные маркёры CD13, CD33 и CD65; маркёры, ассоциированные с моноцитами и гранулоцитами CD14 и CD15; линейные мегакариоцитарные маркёры CD41 и CD61; внутриклеточную миелопероксидазу. Только иммунологическое фенотипирование позволяет установить мегакариоцитарную дифференцировку бластов и провести дифференциальную диагностику с ОЛЛ, М0, М6 вариантами ОМЛ,
метастазами в костный мозг мелкоклеточных злокачественных опухолей
(табл. 3.18). Атипичные клетки экспрессируют антигены CD41a и/или CD42b
и/или CD61. В большинстве случаев на бластных клетках обнаруживается экспрессия миелоидных антигенов CD13, CD33, встречаются линейно неограниченные антигены – HLA-DR, CD38, CD34.
С мегакариобластным лейкозом могут быть связаны аномалии хромосомы 3 – inv(3), t(3;3), t(9;22), трисомия хромосомы 21.
291

Таблица 3.18
Иммунологическая характеристика ОМЛ
Показатели |
М0 |
М1/М2 |
М3 |
М4 |
М5 |
М6 |
М7 |
МПО |
+/– |
+ |
+ |
+ |
–/+ |
–/+ |
– |
CD117 |
+/– |
+ |
+ |
+ |
+/– |
– |
– |
CD13 |
+/– |
+ |
+ |
+ |
+/– |
– |
+/– |
CD14 |
– |
– |
– |
+/– |
+/– |
– |
– |
CD64 |
– |
+/– |
+/– |
+ |
+ |
+/– |
– |
CD15 |
– |
+/– |
–/+ |
+/– |
+ |
– |
– |
CD33 |
+/– |
+/– |
+ |
+ |
+ |
+ |
+ |
CD34 |
+/– |
+/– |
– |
–/+ |
– |
– |
– |
CD41 |
– |
– |
– |
– |
– |
– |
+ |
CD61 |
– |
– |
– |
– |
– |
– |
+ |
Гликофорин A |
– |
– |
– |
– |
– |
+ |
– |
HLA-DR |
+/– |
+ |
– |
+ |
+ |
– |
+ |
- – отрицательная реакция, -/+ – экспрессия антигена отмечается в менее 50% случаев, +/-
– экспрессия антигена отмечается в более 50% случаев, + – положительная реакция
Исследование методом проточной цитофлюорометрии в диагностике острого миелобластного лейкоза существенно в случаях, когда необходима верификация вариантов М0 и М1, а также в диагностике бифенотипического лейкоза. Кроме того, метод позволяет разграничить варианты М0 и М1, а
также варианты с гранулоцитарной дифференцировкой – М2 и М3.
Наибольшую важность приобрели цитогенетические характеристики,
именно они признаны решающими прогностическими факторами.
Цитогенетические изменения в лейкемических клетках обнаруживают у
55-78% взрослых пациентов и у 77-85% детей. Ниже приведено описание наиболее частых и значимых в клиническом отношении хромосомных аберраций при остром миелобластном лейкозе и их прогностическое значение.
Самая частая хромосомная аберрация – t(8;21)(q22;q22), в 90% случаев t(8;21) ассоциирована с вариантом М2, в 10% – с М1. Транслокацию t(8;21)
относят к числу аберраций «благоприятного прогноза». Её обнаруживают у
10-15% детей с острым миелобластным лейкозом.
Транслокация – t(15;17)(q22;ql2) с образованием химерного гена PML-
RARa, составляет 6-12% всех случаев острого миелобластного лейкоза у
292
детей; при варианте М3 она равна 100%. Транскрипт PML-RARa – маркёр лейкемии. У пациентов, достигших ремиссии, его не обнаруживают, а
повторное его выявление во время морфологической ремиссии – предвестник клинического рецидива острого промиелоцитарного лейкоза. Инверсия хромосомы 16 – inv(16)(pl3;q22) – и её вариант t(16;16) характерны для миеломоноцитарного лейкоза с эозинофилией М4Е0, хотя наблюдаются и при других вариантах острого миелобластного лейкоза.
Регион 23 длинного плеча одиннадцатой хромосомы достаточно часто становится участком структурных реаранжировок у детей с острым лейкозом
– как лимфобластным, так и миелобластным. При первичном остром миелобластном лейкозе аномалию llq23 обнаруживают у 6-8% больных, при вторичном – у 85%.
Инверсия inv(3)(q21q26)/t(3;3)(q21;q26) описана при всех вариантах острого миелобластного лейкоза, за исключением M3/M3v и М4Е0. Несмотря на отсутствие связи между определённым FAB-вариантом и инверсией хромосомы 3, у большинства больных в костном мозге обнаруживают общие морфологические признаки: увеличение числа мегакариоцитов и многочисленные микромегакариоциты.
Транслокация t(6;9)(p23;q34) описана более чем у 50 пациентов с острым миелобластным лейкозом. В большинстве случаев это единственная хромосомная аномалия. Несколько чаще t(6;9) выявляют у пациентов с М2 и
М4 вариантами, хотя встречается она при всех формах острого миелобластного лейкоза.
Транслокация t(8;16)(pll;pl3) описана у 30 больных острым миелолейкозом, преимущественно при вариантах М4 и М5. Чаще аномалию обнаруживают у юных пациентов, в том числе у детей до года.
Острые лимфобластные лейкозы. Острые лимфобластные лейкозы
(ОЛЛ) – гетерогенная группа заболеваний, каждое из которых имеет клинические, иммунологические и прогностические особенности. ОЛЛ у детей составляет до 75% гемобластозов и до 25% всех опухолей. Сравнение
293
ФАБ и ВОЗ классификаций ОЛЛ представлено в табл. 3.19. При наличии в костном мозге менее 20% бластных клеток состояние расценивается как лимфобластная лимфома.
|
Таблица 3.19 |
Сравнение ФАБ и ВОЗ классификаций ОЛЛ |
|
|
|
ФАБ-классификация |
ВОЗ-классификация |
>30% бластов |
>20% бластов |
L1/L2 морфология |
Пре-B-ОЛЛ (цитогенетические подгруппы) |
Морфологический вариант различный |
t(9;22)(q34;q11.2) BCR/ABL |
|
t(V;11)(V;q23) MLL реарранжировки |
|
t(1;19)(q23;p13.3) E2A/PBX1 |
|
t(12;21)(p13;q22) |
|
ETV6/RUNX1 (TEL/AML1) |
|
Гиподиплоидия <46 хромосом |
|
Гипердиплоидия >50 хромосом |
|
Пре-T-ОЛЛ (цитогенетические подгруппы |
|
t(V;14)(V;q11–13) |
L3 |
Беркиттоподобный лейкоз |
Цитохимические признаки бластных клеток при ОЛЛ: лимфобласты характеризуются положительной PAS-реакцией, выявляющейся в >3%
клеток в виде мелких или крупных гранул, иногда сливающихся в блоки, и
отрицательной реакцией на МПО и хлорэцетатэстеразу. Реакция на липиды чаще всего отрицательная.
Наиболее информативным и решающим методом диагностики ОЛЛ является проточная цитофлуориметрия с использованием моноклональных антител, которая позволяет установить линейную направленность бластных клеток, а также стадию дифференцировки внутри каждой линии,
диагностировать бифенотипические и билинейные ОЛ. Современная диагностика ОЛЛ должна включать цитогенетическое и молекулярно-
генетическое исследование для идентификации клинически значимых генетических хромосомных аббераций.
Согласно предложению Европейской группы по изучению острых лейкозов все ОЛЛ разделяют на две группы: В- и Т-линейной направленности, которые, в свою очередь, подразделяются на подтипы в
294
зависимости от стадии нормальной дифференцировки лимфоцитов
(табл. 3.20).
Наиболее благоприятным считается В-II (common) вариант ОЛЛ, чаще
всего встречающийся у детей. Неблагоприятными |
прогностическими |
|||||
факторами течения ОЛЛ являются: возраст менее 1 |
|
года, наличие Ph- |
||||
хромосомы, |
раннее |
поражение |
центральной |
нервной |
системы, |
|
распространенность опухолевого процесса, Т-клеточный вариант ОЛЛ.
|
Таблица 3.20 |
|
Иммунологическая классификация ОЛЛ (EGIL, 1995) |
||
|
|
|
Фенотип |
Антигены |
|
В-линейные*: |
– |
|
В-I (про-В) ОЛЛ |
CD19+ и/или CD79a+ и/или CD22+цитоплазматический |
|
В-II (common) ОЛЛ |
CD10+ |
|
В-III (пре-В) ОЛЛ |
Цитоплазматическая μ-цепь+ |
|
В-IV (зрелый-В) ОЛЛ |
Поверхностный IgM моноклональный по κ-или ƛ-типу |
|
|
|
|
|
Большинство случаев Tdt+, HLA-DR+, кроме B-IV, |
|
|
который часто Tdt- |
|
Т-линейные: |
Цитоплазматический или мембранный CD3+ |
|
Т-I (про-Т) ОЛЛ |
CD7+ |
|
T-II (пре-Т) ОЛЛ |
CD2+ и/или CD5+ и/или CD8+ |
|
T-III (кортикальный Т) |
CD1a+ |
|
ОЛЛ |
|
|
T-IV (зрелый Т) ОЛЛ |
Мембранный CD3+,CD1a- |
|
*Позитивные с двумя из трех маркеров
Смешанные острые лейкозы. Острые лейкозы, при которых опухолевые клетки имеют признаки более одной линии дифференцировки,
например, лимфоидной и миелоидной, называются смешанно-линейными,
гибридными, бифенотипическими. Коэкспрессию маркеров различных линий объясняют тем, что лейкемогенез – это не абсолютный блок клеточной дифференцировки, а объединение беспорядка созревания и пролиферации,
дающее возможность экспрессии антигенов, которые в норме отсутствуют.
Описано две категории смешанных ОЛ – ОЛЛ с миелоидно-
ассоциированными антигенами (My+ALL) и ОМЛ с лимфоидно-
ассоциированными антигенами (Ly+AML). Ниже представлены критерии для классификации этих вариантов ОЛ:
295
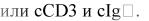
при В-линейном My+ALL бластные клетки должны обязательно экспрессировать:
o CD79a или cIg или CD19 и CD22; o CD3-;
o MPO-;
o CD13, CD15, CD33 или CD65.
при T-линейном My+ALL бластные клетки должны обязательно экспрессировать:
o CD7 и CD3 (поверхностный или цитоплазматический); o CD79a-;
o MPO-;
o CD13, CD15, CD33 или CD65.
при Ly+AML бластные клетки должны обязательно экспрессировать:
o MPO или экспрессия не менее двух других миелоидных
маркеров;
o CD3-;
o CD79a-;
oCD2, CD5, CD7 CD19 CD22 или CD56.
при «истинном» смешанно-линейном лейкозе бластные клетки коэкспрессируют:
o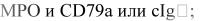
oили MPO и CD3;
o
3.5.3. Миелодиспластические синдромы
Термин миелодиспластические синдромы (МДС) объединяет группу прогрессирующих, необратимых опухолевых заболеваний стволовой кроветворной клетки, характеризующихся длительно существующими цитопениями крови при гиперили нормоклеточном костном мозге,
качественными (диспластическими) изменениями всех миелоидных ростков,
296
предрасположенностью к трансформации в острый нелимфобластный лейкоз. Заболеваемость МДС довольно высокая – в среднем 3,5-7,7 на
100 000 населения в год, в возрастной группе старше 70 лет – около 50 на
100 000. МДС – болезнь пожилых людей: более 80% случаев в возрасте старше 60 лет, не более 10% – в возрасте до 50 лет.
В результате подавления нормального кроветворения, ускоренной гибели и функциональной неполноценности клеток патологического клона развиваются основные клинические проявления заболеваний, объединенных
вгруппу МДС:
анемия (в 85-90% случаев);
нейтропения (у 50% больных, является причиной возникновения инфекционных заболеваний у 10-40%);
тромбоцитопения (приводит к кровоточивости у 25-70% больных);
похудание и лихорадка.
Решающее значение в диагностике МДС имеют данные лабораторных исследований крови и костного мозга.
Диагностическая триада МДС:
хроническая рефрактерная цитопения;
повышенная клеточность костного мозга;
дисплазия одного или нескольких ростков гемопоэза.
Диспластические изменения клеток крови наблюдаются не только при МДС, но и при воздействии ряда лекарственных веществ и токсичных препаратов, проникающего излучения, у лиц, инфицированных ВИЧ, у
больных вирусными гепатитами, туберкулезом, при лейкозах, дефиците витамина В12 и фолиевой кислоты. Обычно при МДС костный мозг бывает гиперили нормоклеточным, однако примерно в 10% случаев наблюдается его гипоплазия, что требует дифференциальной диагностики с апластической анемией. Гистологическое исследование трепанобиоптата костного мозга обязательно при диагностике МДС, поскольку позволяет достоверно оценить
297
клеточность костного мозга, а также выявить характерные особенности, в
частности атипичную локализацию миелоидных предшественников (АЛМП)
в центральных отделах костномозговых полостей, а не вблизи костных балок.
ФАБ-классификация была разработана в начале 80-х годов прошлого века, основным критерием в данной классификации являлось процентное содержание бластных клеток в костном мозге, при этом процентное содержание этих клеток менее 2% считалось нормальным для здорового костного мозга. Согласно ФАБ-классификации различаются следующие пять подтипов МДС:
рефрактерная анемия (РА);
рефрактерная анемия с кольцевыми сидеробластами (РАКС);
рефрактерная анемия с избытком бластов (РАИБ);
рефрактерная анемия с избытком бластов в трансформации (РАИБ-Т);
хронический миеломоноцитарный лейкоз (ХММЛ).
Система классификации ВОЗ для МДС у взрослых больных сохраняет некоторые элементы системы ФАБ-классификации и расширяет категории подтипов МДС. Основные характеристики подтипов МДС, различаемых системой классификации ВОЗ, приведены в табл. 3.21.
|
|
|
Таблица 3.21 |
|
|
ВОЗ-классификация МДС |
|||
|
Подтип МДС |
|
Характеристика |
|
|
|
|
||
|
|
|
|
|
|
|
|
Минимальная дисплазия в одном типе клеток |
|
|
Рефрактерная анемия (РА). |
|
крови (красных кровяных тельцах или |
|
|
|
|
эритроцитах) и < 5% бластов в костном мозге |
|
|
Рефрактерная анемия с кольцевыми |
|
Дисплазия только эритроидного ростка и > 15% |
|
|
сидеробластами (РАКС). |
|
кольцевых сидеробластов |
|
|
|
|
Дисплазия (> 10%) в двух или трех типах клеток |
|
|
Рефрактерная цитопения с |
|
крови, < 5% бластов и < 15% кольцевых |
|
|
мультилинейной дисплазией |
|
сидеробластов в костном мозге (количество |
|
|
(РЦМД). |
|
кольцевых сидеробластов > 15% называется |
|
|
|
|
РЦМД-КС) |
|
|
Рефрактерная анемия с избытком |
|
РАИБ 1 – количество бластов в костном мозге от |
|
|
|
5% до 9% |
|
|
|
бластов (РАИБ). |
|
|
|
|
|
РАИБ 2 – количество бластов в костном мозге от |
|
|
|
|
|
|
|
|
|
298 |
|
|
|
10% до 19% |
Миелодиспластический |
Дисплазия при наличии характеристик, обычно |
синдром/миелопролиферативное |
присущих миелопролиферативному заболеванию; |
заболевание (МДС/МПЗ). |
включает ХММЛ |
|
Больные, у которых отсутствуют хромосомные |
5q- (5q минус) синдром. |
аномалии, за исключением делеции в длинном |
|
плече 5-й хромосомы |
|
Включает больных с цитопенией в одном типе |
|
клеток крови, кроме анемии (то есть, с |
Неклассифицируемый МДС. |
нейтропенией или тромбопенией) и какими-либо |
|
необычными характеристиками (например, |
|
фиброзом костного мозга) |
Рефрактерная анемия (РА) с однолинейной дисплазией. Больные этой категории страдают анемией, которая не отвечает на лечение препаратами железа или витаминами, то есть является рефрактерной по отношению к такому лечению. Анемия может сопровождаться тромбоцитопенией и нейтропенией легкой или средней тяжести.
Рефрактерная анемия с кольцевыми сидеробластами (РАКС). У
больных с данным видом анемии отмечается дисплазия только в эритроидном ряду. Сидеробласты – клетки эритроидного ряда, содержащие гранулы железа; кольцевые сидеробласты являются аномальными.
Рефрактерная анемия с кольцевыми сидеробластами (РАКС) считается наиболее доброкачественным подтипом в системе классификации ВОЗ.
Рефрактерная цитопения с мультилинейной дисплазией (РЦМД). В
эту категорию включаются больные, страдающие рефрактерной цитопенией
(устойчивым низким количеством клеток крови какого-либо типа, например,
рефрактерной нейтропенией или рефрактерной тромбоцитопенией) и
имеющие минимальную дисплазию, по меньшей мере, в двух типах клеток крови, а также количество бластов 5% и менее, либо количество кольцевых сидеробластов менее 15%. Если количество кольцевых сидеробластов у больного с РЦМД превышает 15%, ставится диагноз РСМД-КС.
Рефрактерная анемия с избытком бластов (РАИБ). Эта категория делится на две подкатегории, которые различаются количеством бластов в
299
костном мозге. У больных, имеющих РАИБ-1, количество бластов составляет от 5% до 9%, а у больных с РАИБ-2 – от 10% до 19%.
Миелодиспластический синдром / миелопролиферативное заболевание (МДС/МПЗ). Больные с МДС/МПЗ включают тех, которые страдают хроническим миеломоноцитарным лейкозом (ХММЛ), который является отдельной категорией в системе ФАБ-классификации.
5q- (5q минус) синдром. Синдром 5q-, в настоящее время выделяемый в отдельный подтип МДС, впервые был описан более 30 лет тому назад.
Название этого синдрома связано с хромосомной аномалией (делецией) в
длинном плече (q) хромосомы 5. Делеция в пределах длинного плеча хромосомы 5 является единственной хромосомной аномалией у больных МДС с диагнозом «синдром 5q-».Обычно этот синдром наблюдается у женщин среднего возраста, как правило, в возрасте 65 лет и сопровождается анемией легкой или средней тяжести, низким количеством лейкоцитов
(лейкопения) и зачастую нормальным или повышенным количеством тромбоцитов. Синдром 5qхарактеризуется благоприятным прогнозом,
длительностью жизни более пяти лет со времени постановки диагноза.
Неклассифицируемый МДС. Данная категория включает больных с цитопенией в одном типе клеток крови (например, тромбопенией или нейтропенией) и какими-либо необычными характеристиками (например,
фиброзом костного мозга). Этот синдром встречается относительно редко – не более 1%-2% всех случаев МДС.
3.5.4. Миелопролиферативные заболевания. Классификация
миелопроферативных заболеваний
Хронические миелопролиферативные заболевания – клональные опухоли, развивающиеся из стволовой кроветворной клетки,
характеризующиеся пролиферацией в костном мозге одного или более ростков миелоидной линии (гранулоцитарного, эритроидного,
мегакариоцитарного). Пролиферация клеток сопровождается относительно
300