

Маслов Лабораторныы практикум Компютерное моделирование графена 2015
.pdf3.Hamada N., Sawada S., Oshiyama A. New one-dimensional conductors: Graphitic microtubules // Phys. Rev. Lett. 1992. V. 68. P. 1579.
4.Kienle D., Cerda J.I., Ghosh A.W. Extended Hückel theory for band structure, chemistry, and transport. I. Carbon nanotubes // J. Appl. Phys. 2006. V. 100. P. 043714.
5.Smith B.W., Monthioux M., Luzzi D.E. Encapsulated C60 in carbon nanotubes // Nature. 1998. V. 396. P. 323.
6.Ivanovskaya V.V., Makurin Yu.N., Ivanovskii A.L. Fullerene peapods and related nanomaterials: synthesis, structure and electronic properties. In: Nanostructures Novel Architectures. Nova Sci. Publ., N.Y. 2005. Chap. 2. P. 9.
7.Monthioux M. Filling single-wall carbon nanotubes // Carbon. 2002. V. 40. P. 1809.
8.Ивановский А.Л. Фуллерены и родственные наночастицы, инкапсулированные в нанотрубки: синтез, свойства и моделирование новых гибридных наноструктур // ЖНХ. 2003. Т. 48. С. 945.
31

ПРИЛОЖЕНИЕ 1
ВИЗУАЛИЗАЦИЯ ДАННЫХ
Обычно при работе с программами, в которых реализуется численный расчет структуры и динамики графеновых фрагментов или других типов наноструктур, на начальном этапе формирования исходных расчетных данных бывает необходимо представить в наглядной форме имеющиеся данные. Такая необходимость существует в тех случаях, когда нужно отслеживать особенности хода процессов молекулярно-динамического моделирования. Для наглядного представления данных задач лабораторных работ 1 – 5 настоящего пособия предлагается использовать программу визуализации ClustVis, созданную в качестве прикладной программы в ходе исследований на кафедре физики конденсированных сред.
Общая характеристика молекулярного визуализатора ClustVis
Молекулярный визуализатор ClustVis предназначен для построения трехмерных структурных моделей атомных кластеров и молекул и анализа их геометрии. В визуализатор встроен дополнительный модуль, реализующий алгоритм сильной связи NTBM, что позволяет проводить квантово-химическую оптимизацию структуры молекул и наблюдать результаты расчета в режиме реального времени. Простота интерфейса и достаточно мощный функционал выделяют программу ClustVis среди других программных пакетов, предназначенных для визуализации молекул. ClustVis работает под управлением операционной системы Windows 7, распространяется бесплатно и имеет открытый программный код. Ниже приведено подробное описание интерфейса и возможностей визуализатора
ClustVis.
Структура входных файлов
Входной файл представляет собой обычный текстовый документ, состоящий из нескольких строк. Каждая строка содержит де-
32
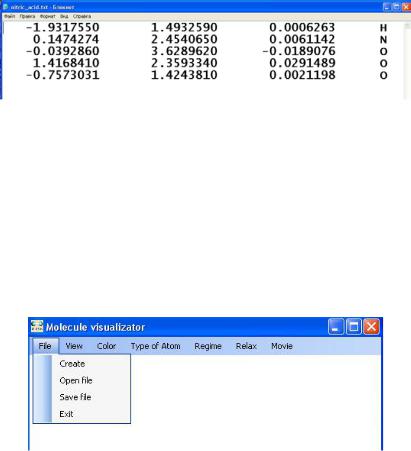
картовы координаты атома, входящего в молекулу, и химический символ соответствующего элемента. Координаты приводятся в ангстремах, целая и дробная часть числа разделяются точкой. Координаты отделяются друг от друга, а также от символа химического элемента, одним или несколькими пробелами. Порядок, в котором указаны атомы, не влияет на построение изображения. Пример входного файла, описывающего молекулу азотной кислоты, приведен на рис. П.1.1.
Рис. П.1.1. Пример входного файла, описывающего молекулу азотной кислоты
Интерфейс ввода-вывода
Чтобы визуализировать структуру молекулы, содержащейся во входном файле, необходимо в меню «File» выбрать пункт «Open file», а затем в открывшемся окне выбрать нужный файл. Чтобы сохранить представленную на экране молекулу в виде файла, необходимо в меню «File» выбрать пункт «Save file», а затем в откры в- шемся окне ввести имя нужного файла. Соответствующее меню представлено на рис. П.1.2.
Рис. П.1.2. Интерфейс ввода-вывода
33
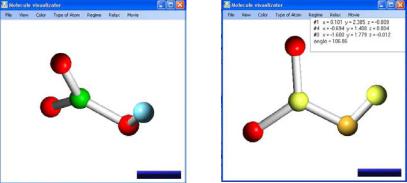
Анализ структуры молекулы
Молекула изображается на экране при помощи представления «атом–связь». Атомы условно изображаются шарами разных цветов, а связи между ними – цилиндрами. Связь изображается только в том случае, если соответствующие атомы находятся достаточно близко друг к другу (расстояние не более 1,8 Å), при этом количество связей каждого атома не превышает его валентности. Для правильной оценки масштаба внизу изображается метка, длина которой составляет 1 Å (рис. П.1.3). Изменение масштаба осуществляется при помощи колеса прокрутки компьютерной «мыши».
Рис. П.1.3. Анализ структуры молекулы азотной кислоты
ClustVis позволяет визуально исследовать структуру молекулы, «рассматривая» ее с разных сторон. Для того чтобы повернуть изображение, представленное на экране, необходимо перетащить указатель компьютерной мыши по области изображения.
Чтобы вывести на экран декартовы координаты отдельного атома, нужно выделить его, щелкнув по нему левой клавишей компьютерной «мыши». При последовательном выделении двух атомов выводится также расстояние между ними (в ангстремах), трех атомов – величина образуемого ими угла (в градусах), четырех атомов – величина угла между векторами, соединяющими первый со вторым и третий с четвертым атомами (в градусах). Можно также вывести на экран номера атомов (в том порядке, в каком они
34

представлены во входном файле). Для этого необходимо в меню «View» выбрать пункт «Numbers». Примеры изображений, получающихся в результате работы визуализатора, приведены на рис. П.1.3.
При проведении квантово-химических расчетов иногда бывает удобно визуально выделить группу атомов, изменив их цвет и/или прозрачность. Это можно сделать, выбрав в меню «View» пункт «Set color of atoms». Соответствующий интерфейс и результат применения этого инструмента (на примере кластера графена) представлены на рис. П.1.4.
Рис. П.1.4. Выделение цветом отдельной группы атомов в кластере графена
Молекулярный конструктор
Визуализатор ClustVis позволяет не только анализировать структуру молекулы, но и модифицировать ее, добавляя, удаляя или перемещая атомы. Для того чтобы добавить новый атом, необходимо в меню «Regime» выбрать пункт «Add and Delete». Затем в меню «Type of Atom» нужно выбрать символ химического элемента, атом которого планируется добавить в систему. После этого атом создается в любой точке области изображения при помощи левой клавиши компьютерной «мыши». Чтобы удалить атом из системы, следует навести на него указатель компьютерной «мыши» и щелкнуть правой клавишей.
35
Для перемещения атомов следует в меню «Regime» выбрать пункт «Rotate and Move». В этом режиме можно перетащить любой атом в любое место при помощи компьютерной «мыши». Все изменения, произошедшие с рассматриваемой структурой в результате добавления, удаления и перемещения атомов, можно сохранить при помощи пункта «Save file» в меню «File». Кроме того, можно создать новый «чистый» файл для конструирования новой молекулярной структуры. Для этого необходимо в меню «File» выбрать пункт «Create».
Молекулярная анимация
ClustVis позволяет также визуализировать молекулярные процессы при помощи анимационных фильмов. Каждый «кадр» такого фильма описывает структуру молекулы в определенный момент времени. Кадры воспроизводятся на экране один за другим, что создает иллюзию «движения» атомов. Такое визуальное представление молекулярных процессов часто используется для анализа результатов молекулярно-динамического моделирования.
Структура входного анимационного файла очень похожа на структуру обычного входного файла, описанного в соответствующем разделе. Он представляет собой обычный текстовый документ, в первой строчке которого записано число, определяющее количество атомов в структуре. Далее последовательно описываются «кадры» анимационного фильма в том порядке, в котором они будут воспроизводиться. Количество кадров не ограничено. Пример подобного файла, содержащего всего три кадра и имитирующего разложение азотной кислоты, представлен на рис. П.1.5.
Для управления воспроизведением анимационного фильма разработана специальная панель, включающая стандартные инструменты для работы с видео: полосу прокрутки, кнопки для начала и остановки воспроизведения, а также для управления его скоростью (рис. П.1.6). С каждым кадром анимационного фильма можно работать так же, как с отдельной структурой: сохранять, модифицировать, вращать; добавлять, удалять и перемещать атомы.
36

Рис. П.1.5. Пример входного файла анимационного фильма, содержащего
три кадра и имитирующего разложение азотной кислоты
Рис. П.1.6. Панель для управления воспроизведением анимационного фильма
Структурная оптимизация
Любая молекулярная система, предоставленная самой себе и охлажденная до абсолютного нуля, меняет свою структуру, переходя в состояние, отвечающее глобальному или локальному минимуму потенциальной энергии. Визуализатор ClustVis позволяет моделировать этот процесс, называемый геометрической оптими-
37

зацией, благодаря специальному встроенному модулю, реализующему неортогональный алгоритм сильной связи NTBM. Геометрическая оптимизация визуализируется при помощи молекулярной анимации.
Чтобы оптимизировать структуру, необходимо в меню «Relax» выбрать пункт «Start relaxing dll». На рис. П.1. 7. приведена структура бензола до и после оптимизации.
а б
Рис. П.1.7. Структура молекулы бензола до (а) и после (б) оптимизации
Таким образом, ClustVis предоставляет собой инструментарий не только для визуализации молекулярных структур и создание анимационных фильмов (например, по распаду или изомеризации), но и проведения полуэмпирических расчетов геометрических характеристик и структурной оптимизации выбранной системы.
Рекомендуемая литература
1. Pischulina A.Yu., Shostachenko S.A., Katin K.P., Prudkovskiy V.S., Maslov M.M., Ryzhuk R.V., Kargin N.I. ClustVis1: A new software package for visualization of atomic clusters and molecules // Scientific Visualization. 2015. V. 7. P. 30–37.
38

ПРИЛОЖЕНИЕ 2
НЕОРТОГОНАЛЬНАЯ МОДЕЛЬ СИЛЬНОЙ СВЯЗИ ДЛЯ РАСЧЕТА H–C–N–O СОЕДИНЕНИЙ
Лабораторные работы 1 – 5, представленные в настоящем практикуме, разработаны с непосредственным использованием неортогонального потенциала сильной связи для H–C–N–O соединений, являющегося оригинальной разработкой авторского коллектива (см. ntbm.info). Так, геометрия графена и наноструктур на его основе, предлагаемых студентам для исследования в лабораторных работах 1 – 5, определялась в рамках NTBM. Поскольку выполнение лабораторных работ требует глубокого понимания физических основ компьютерного моделирования наносистем, в настоящем приложении на примере неортогонального потенциала сильной связи для расчета H–C–N–O соединений приводится детальное описание модели сильной связи как эффективной методики моделирования различных наноструктур.
Описание модели
При заданных значениях координат всех атомов {Ri } полная потенциальная энергия системы в модели сильной связи
E = Eel + Erep . |
(П.2.1) |
Здесь |
|
Eel = ∑ en − |
(П.2.2) |
n,σ(occup) |
|
квантово-механическая электронная составляющая |
E, равная сум- |
ме энергий εn занятых одноэлектронных состояний (в отсутствие
магнитного поля εn не зависит от проекции спина σ = |
↑ или ↓); |
Erep = ∑∑Φ(Rij ) − |
(П.2.3) |
i j>i |
|
39 |
|

«классическая» составляющая E, равная сумме двухчастичных энергий отталкивания ионов
Φ(Rij )= Φ(Ri −R j ).
Энергетический спектр {εn } находится из стационарного уравнения Шредингера
|
(П.2.4) |
ΗΨn (r)=εnΨn (r) |
|
путем разложения собственных функций |
|
Ψn (r)= ∑Cinαφiα (r) |
(П.2.5) |
i,α |
|
по неортогональному набору атомных орбиталей {φiα (r)}, где i –
номер атома; α – тип атомной орбитали этого атома (учитываются 1S орбитали атомов водорода и 2S, 2Px, 2Py, 2Pz орбитали атомов углерода, азота и кислорода). После подстановки (П.2.5) в (П.2.4),
домножения слева на φ*jβ (r)и интегрирования по r уравнения на собственные значения принимают вид
∑( |
H jβ −ε |
S jβ Cn |
= 0, |
(П.2.6) |
iα n |
iα ) iα |
|
|
i,α
где
Siαjβ = ∫drφ*jβ (r)φiα (r) −
матричные элементы перекрытия неортогональных (в общем случае) атомных орбиталей,
jβ |
* |
|
Hiα |
= ∫drφjβ (r)Hφiα (r) − |
|
матричные элементы гамильтониана системы (заметим, что в ортогональных моделях сильной связи Siαjβ = δij δαβ ).
Таким образом, одноэлектронное уравнение Шредингера (П.2.4) сводится к обобщенной задаче на собственные значения (П.2.6), которая решается численно. Число уравнений в системе (П.2.6) равно полному числу рассматриваемых атомных орбиталей, а именно:
40