

10. Кислоты и основания (Бренстед, Льюис). Сопряженные кислоты и основания. Кислотно-основные равновесия. Константа кислотной ионизации и ее показатель (рКа).
Ответ.
Для оценки кислотности и основности
органических соединений в современной
органической химии используют две
теории — протонную (протолитическую)
теорию Брёнстеда и электронную теорию
Льюиса. Согласно теории Брёнстеда
кислотой называют любое вещество,
способное отдавать протон (донор
протона), а основанием — вещество,
способное присоединять протон (акцептор
протона). Отсюда теория получила название
«протонной» или «протолитической». Для
взаимодействия с протоном основание
должно иметь неподеленную пару электронов
или π-молекулярную орбиталь. Кислотность
и основность являются относительными
свойствами вещества. Кислотный характер
может проявляться лишь в присутствии
основания, и, наоборот, основный характер
— только в присутствии кислоты. В целом
кислотно-основный процесс состоит в
переносе протона от кислоты к основанию
и может быть представлен следующей
схемой:
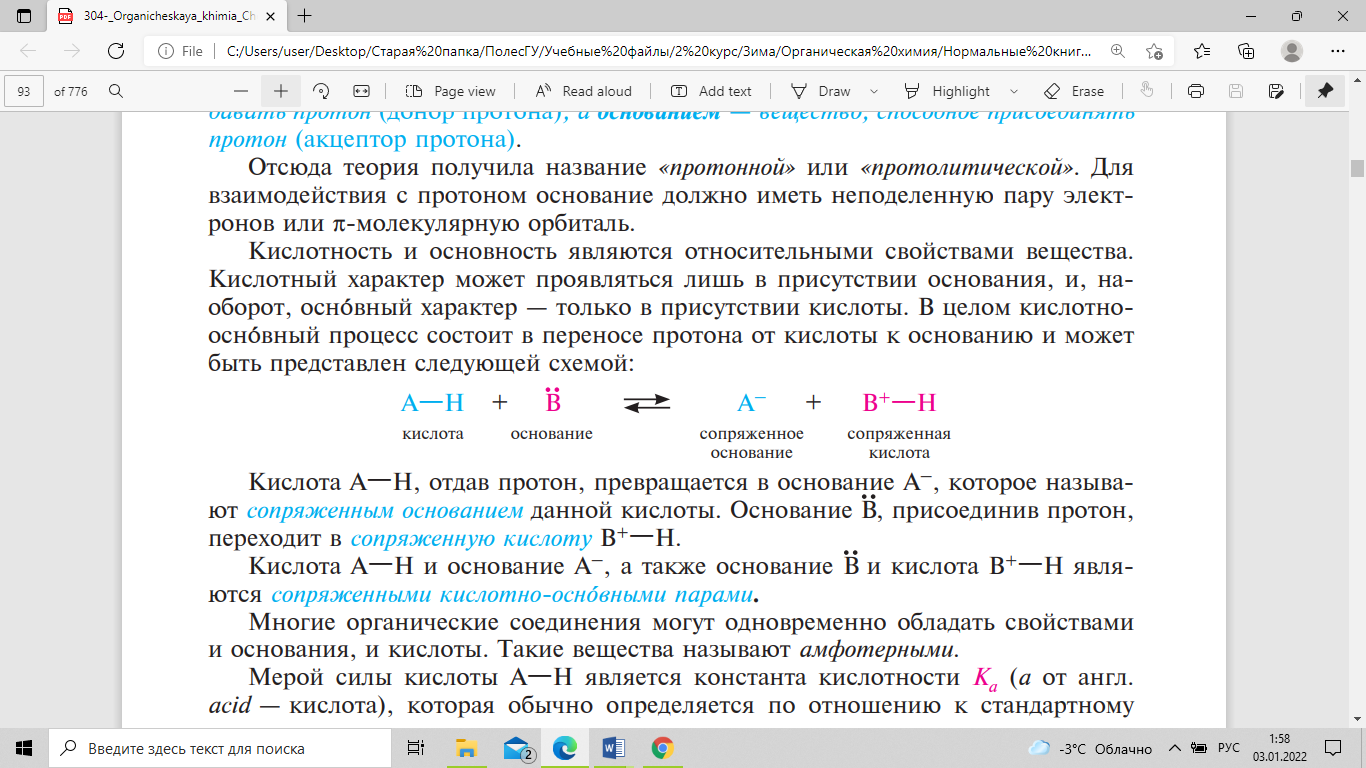 .
Кислота А—Н, отдав протон, превращается
в основание А–, которое называют
сопряженным
основанием данной кислоты. Основание
В, присоединив протон, переходит в
сопряженную кислоту В+—Н. Кислота А—Н
и основание А–, а также основание В и
кислота В+—Н являются сопряженными
кислотно-основными парами. Мерой силы
кислоты А—Н является константа
кислотности Kа, которая обычно определяется
по отношению к стандартному основанию
— воде. В сильно разбавленном растворе
Ка рассчитывают по формуле
.
Кислота А—Н, отдав протон, превращается
в основание А–, которое называют
сопряженным
основанием данной кислоты. Основание
В, присоединив протон, переходит в
сопряженную кислоту В+—Н. Кислота А—Н
и основание А–, а также основание В и
кислота В+—Н являются сопряженными
кислотно-основными парами. Мерой силы
кислоты А—Н является константа
кислотности Kа, которая обычно определяется
по отношению к стандартному основанию
— воде. В сильно разбавленном растворе
Ка рассчитывают по формуле
![]() Чем
больше значение Kа, тем сильнее кислота.
Как правило, константы кислотности
очень малы. Например, для уксусной
кислоты Kа при 25 °С равна 1,76·10–5.
Оперировать такими малыми числами
неудобно, поэтому в практической работе
чаще пользуются величинами рKа. рKа =
–lgKa. Так, рKа уксусной кислоты равна
4,75. Чем меньше значение рKа,
тем сильнее кислота. Подобно кислотам,
силу оснований количественно выражают
константой основности Кb (от англ. base —
основание). Константа основности
основания В в воде определяется из
равновесия:
Чем
больше значение Kа, тем сильнее кислота.
Как правило, константы кислотности
очень малы. Например, для уксусной
кислоты Kа при 25 °С равна 1,76·10–5.
Оперировать такими малыми числами
неудобно, поэтому в практической работе
чаще пользуются величинами рKа. рKа =
–lgKa. Так, рKа уксусной кислоты равна
4,75. Чем меньше значение рKа,
тем сильнее кислота. Подобно кислотам,
силу оснований количественно выражают
константой основности Кb (от англ. base —
основание). Константа основности
основания В в воде определяется из
равновесия:
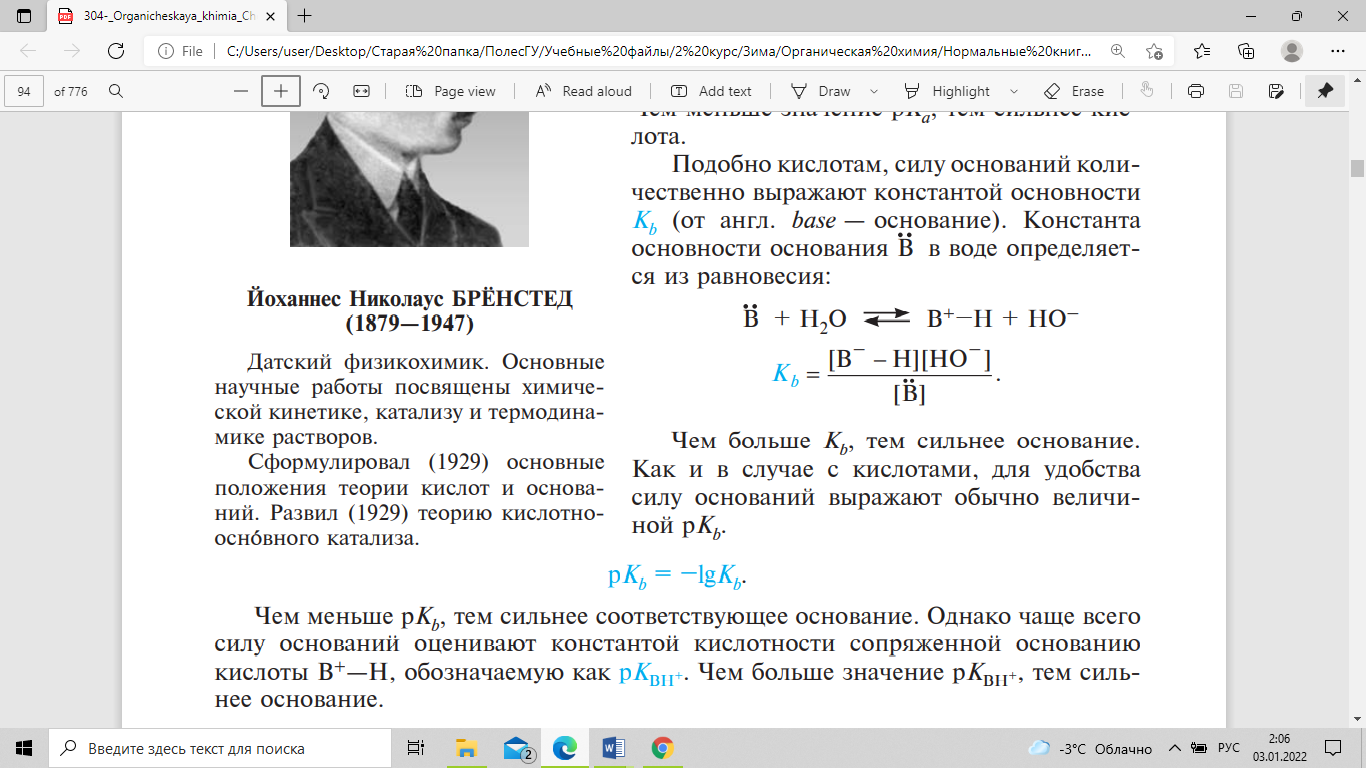 Чем
больше Kb, тем сильнее основание. Для
удобства силу оснований выражают
величиной рKb. рКb = –lgKb. Чем меньше рKb,
тем сильнее соответствующее основание.
Однако чаще всего силу оснований
оценивают константой кислотности
сопряженной основанию кислоты В+—Н,
обозначаемую как рKВН+. Чем больше
значение рKВН+, тем сильнее основание.
Г. Н. Льюис в 1923 году предложил электронную
теорию кислот и оснований, которая не
противоречит Брёнстеду, но является
более общей. Согласно теории Льюиса
основание — любая частица (атом, молекула
или анион), способная отдавать электронную
пару для образования ковалентной связи,
а кислота — любая частица (атом, молекула,
катион), способная принимать пару
электронов с образованием ковалентной
связи. Основание по теории Льюиса
является донором, а кислота — акцептором
пары электронов. Из приведенного
определения видно, что основания Льюиса
тождественны основаниям Брёнстеда.
Однако кислоты Льюиса охватывают более
широкий круг органических соединений.
Кислотой Льюиса считается любая частица,
имеющая вакантную орбиталь. Если в
теории Брёнстеда кислота — это донор
протона, то согласно теории Льюиса сам
протон является кислотой, поскольку
имеет вакантную орбиталь. В представлении
электронной теории кислота Брёнстеда
является соединением, которое образует
кислоту Льюиса. Поэтому согласно теории
Льюиса к кислотам относят не только
соединения, отщепляющие протон (протонные
кислоты), но и другие вещества, имеющие
вакантную орбиталь и способные принимать
пару электронов (апротонные кислоты).
Кислотами Льюиса являются такие
соединения, как BF3, AlCl3, SbCl3, ZnCl2, HgCl2 и др.
Кислотно-основный процесс по Льюису
состоит в образовании ковалентной связи
между основанием и кислотой за счет
электронной пары основания и вакантной
орбитали кислоты. Так, основания Льюиса,
имеющие неподеленные пары электронов,
образуют с кислотами Льюиса n-комплексы:
Чем
больше Kb, тем сильнее основание. Для
удобства силу оснований выражают
величиной рKb. рКb = –lgKb. Чем меньше рKb,
тем сильнее соответствующее основание.
Однако чаще всего силу оснований
оценивают константой кислотности
сопряженной основанию кислоты В+—Н,
обозначаемую как рKВН+. Чем больше
значение рKВН+, тем сильнее основание.
Г. Н. Льюис в 1923 году предложил электронную
теорию кислот и оснований, которая не
противоречит Брёнстеду, но является
более общей. Согласно теории Льюиса
основание — любая частица (атом, молекула
или анион), способная отдавать электронную
пару для образования ковалентной связи,
а кислота — любая частица (атом, молекула,
катион), способная принимать пару
электронов с образованием ковалентной
связи. Основание по теории Льюиса
является донором, а кислота — акцептором
пары электронов. Из приведенного
определения видно, что основания Льюиса
тождественны основаниям Брёнстеда.
Однако кислоты Льюиса охватывают более
широкий круг органических соединений.
Кислотой Льюиса считается любая частица,
имеющая вакантную орбиталь. Если в
теории Брёнстеда кислота — это донор
протона, то согласно теории Льюиса сам
протон является кислотой, поскольку
имеет вакантную орбиталь. В представлении
электронной теории кислота Брёнстеда
является соединением, которое образует
кислоту Льюиса. Поэтому согласно теории
Льюиса к кислотам относят не только
соединения, отщепляющие протон (протонные
кислоты), но и другие вещества, имеющие
вакантную орбиталь и способные принимать
пару электронов (апротонные кислоты).
Кислотами Льюиса являются такие
соединения, как BF3, AlCl3, SbCl3, ZnCl2, HgCl2 и др.
Кислотно-основный процесс по Льюису
состоит в образовании ковалентной связи
между основанием и кислотой за счет
электронной пары основания и вакантной
орбитали кислоты. Так, основания Льюиса,
имеющие неподеленные пары электронов,
образуют с кислотами Льюиса n-комплексы:
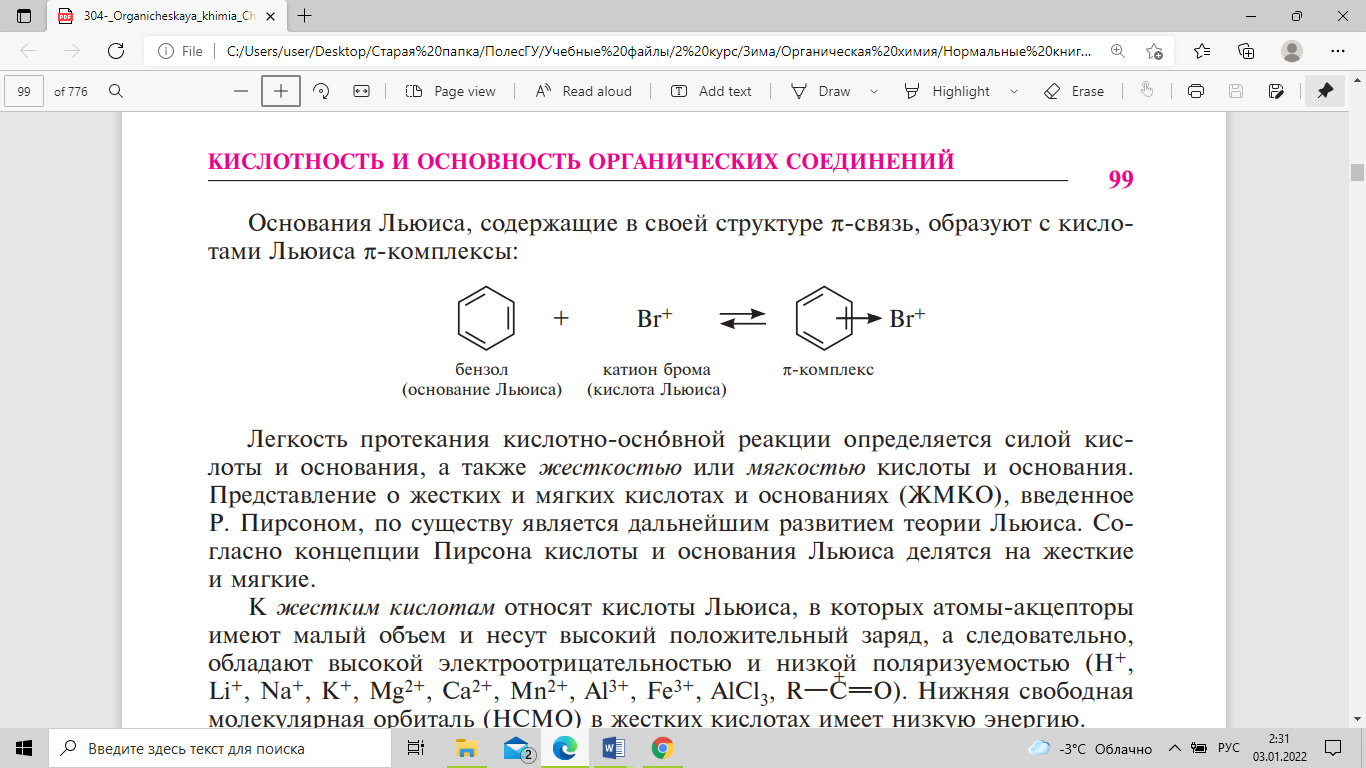
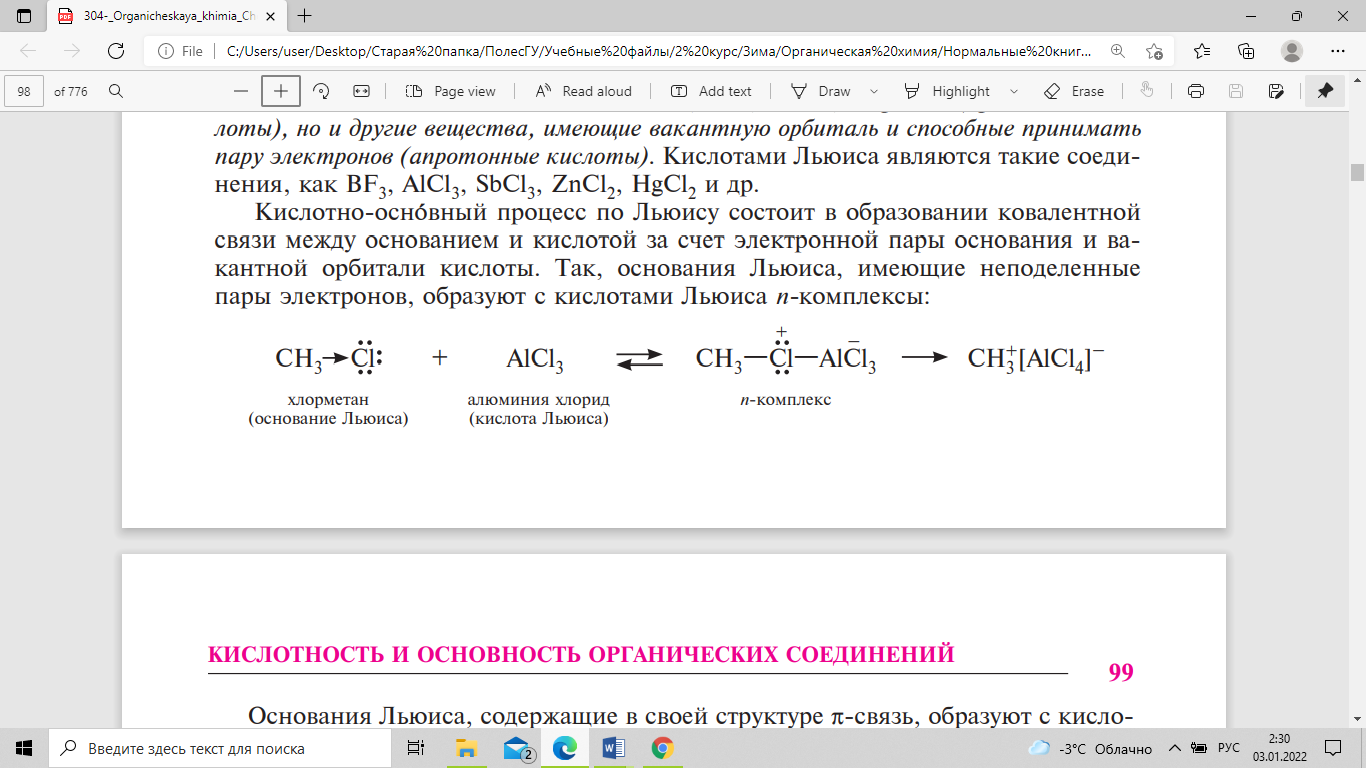 Основания
Льюиса, содержащие в своей структуре
π-связь, образуют с кислотами Льюиса
π-комплексы:
Основания
Льюиса, содержащие в своей структуре
π-связь, образуют с кислотами Льюиса
π-комплексы:
 Лёгкость
протекания кислотно-основной реакции
определяется силой кислоты и основания,
а также жесткостью или мягкостью кислоты
и основания.
Лёгкость
протекания кислотно-основной реакции
определяется силой кислоты и основания,
а также жесткостью или мягкостью кислоты
и основания.