

35. Влияние заместителей в бензольном кольце на изомерный состав продуктов и скорость реакции. Активирующие и дезактивирующие заместители. Орто-, пара- и мета-ориентанты.
Ответ.
В молекуле незамещенного бензола
электронная плотность распределена
равномерно, поэтому электрофильный
реагент может атаковать в равной степени
любой из шести атомов углерода. Если в
бензольном кольце содержится какой-либо
заместитель, то под его влиянием
происходит перераспределение π-электронной
плотности цикла и новая группа вступает
уже в определенные положения по отношению
к имеющемуся заместителю. В реакциях
электрофильного замещения монозамещенных
бензола, в зависимости от электронной
природы заместителя, вступающая группа
может занимать преимущественно орто-,
мета- или пара-положения, а реакция
соответственно протекать быстрее или
медленнее, чем с незамещенным бензолом.
По влиянию на направление реакций
электрофильного замещения и реакционную
способность бензольного кольца
заместители можно разделить на две
группы — заместители I рода (орто-,
пара-ориентанты) и заместители II рода
(мета-ориентанты). К заместителям I рода
относятся атомы и атомные группы,
проявляющие положительный индуктивный
или положительный мезомерный эффекты:
—O–, —NR2, —NHR,
—NH2,
—OH,
—OR,
—NHCOR,
—OCOR,
—SR,
—F,
—Cl,
—Br,
—I, —Alk и др. Заместители I рода (за
исключением галогенов) увеличивают
электронную плотность в бензольном
кольце и тем самым активируют его в
реакциях электрофильного замещения.
Заместители I рода направляют замещение
преимущественно в орто- и параположения.
К заместителям II рода относятся группы,
проявляющие отрицательный индуктивный
или отрицательный мезомерный эффекты:
—NO2,
—SO3H,
—CN,
—CHO,
—COR,
—COOH,
—COOR,
—CONH2,
—CCl3
и др. Заместители II рода уменьшают
электронную плотность в бензольном
кольце и снижают скорость реакции
электрофильного замещения по сравнению
с незамещенным бензолом. Заместители
II рода направляют замещение преимущественно
в мета-положение. Ориентация замещения
не является абсолютной, а свидетельствует
лишь о предпочтительном направлении
реакции с преобладающим образованием
того или иного изомера. Так, при нитровании
нитробензола образуется 93 % мета-, 6 %
орто- и 1 % парадинитробензола. Механизм
влияния заместителей в бензольном
кольце на направление и скорость реакций
электрофильного замещения можно
объяснить с учетом электронных эффектов,
которые играют существенную роль как
в распределении электронной плотности
в стационарном состоянии молекулы
(статический фактор), так и в стабилизации
образующихся в процессе реакции
σ-комплексов (динамический фактор).
Заместители I рода (кроме галогенов) за
счет +I-или +М-эффекта проявляют
электронодонорные свойства. Они повышают
электронную плотность на всех атомах
углерода бензольного кольца, но в большей
степени на углеродных атомах в орто- и
пара-положениях (статический фактор).
Это является причиной облегчения
электрофильного замещения в сравнении
с реакциями SE у незамещенного бензола
и преимущественной атаки электрофильной
частицей орто- и пара-положений.
Заместители II рода, наоборот, за счет
–I- или –М-эффектов проявляют
электроноакцепторные свойства, вызывая
общее уменьшение электронной плотности
в бензольном кольце, но в большей степени
это влияние сказывается в ортои
пара-положениях. Поэтому они затрудняют
реакции электрофильного замещения
вообще и особенно с участием орто- и
пара-положений. В результате замещение
протекает преимущественно в мета-положении.
Наряду со статическим фактором
существенное, а в некоторых случаях
решающее влияние на направление
электрофильного замещения оказывает
динамический фактор. Его сущность
определяется влиянием имеющегося в
бензольном кольце заместителя на
устойчивость образующегося в момент
реакции того или иного σ-комплекса. Из
всех возможных σ-комплексов для молекулы
энергетически более выгодны те, в которых
имеется возможность дополнительной
делокализации положительного заряда
за счет заместителя. Эти σ-комплексы
обладают меньшей энергией, а следовательно,
они более устойчивы и поэтому их
образование в ходе реакции будет более
предпочтительным. В реакциях SЕ заместители
I рода вследствие своих электронодонорных
свойств повышают устойчивость всех
σ-комплексов по сравнению с незамещенным
бензолом и, таким образом, увеличивают
скорость реакции, но в большей степени
они стабилизируют σ-комплексы, отвечающие
продуктам орто- и паразамещения. Например,
при нитровании метоксибензола каждый
из σ-комплексов, образующихся в результате
орто-, мета- и пара-замещения, стабилизирован
за счет делокализации положительного
заряда между атомами углерода бензольного
кольца. Но в σ-комплексах при орто- и
пара-замещении положительный заряд
может быть дополнительно делокализован
с участием неподеленной пары электронов
атома кислорода метоксигруппы. Поэтому
образование их в ходе реакции более
предпочтительно. В результате образуются
преимущественно продукты орто- и
пара-замещения. Заместители ІІ рода
вследствие своих электроноакцепторных
свойств дестабилизируют в той или иной
мере все три возможных σ-комплекса и
тем самым затрудняют электрофильное
замещение в сравнении с незамещенным
бензолом. Однако σ-комплекс в мета-положении
дестабилизируется в меньшей степени,
чем σ-комплекс в орто- и пара-положениях.
Так, при нитровании нитробензола возможно
образование следующих σ-комплексов:

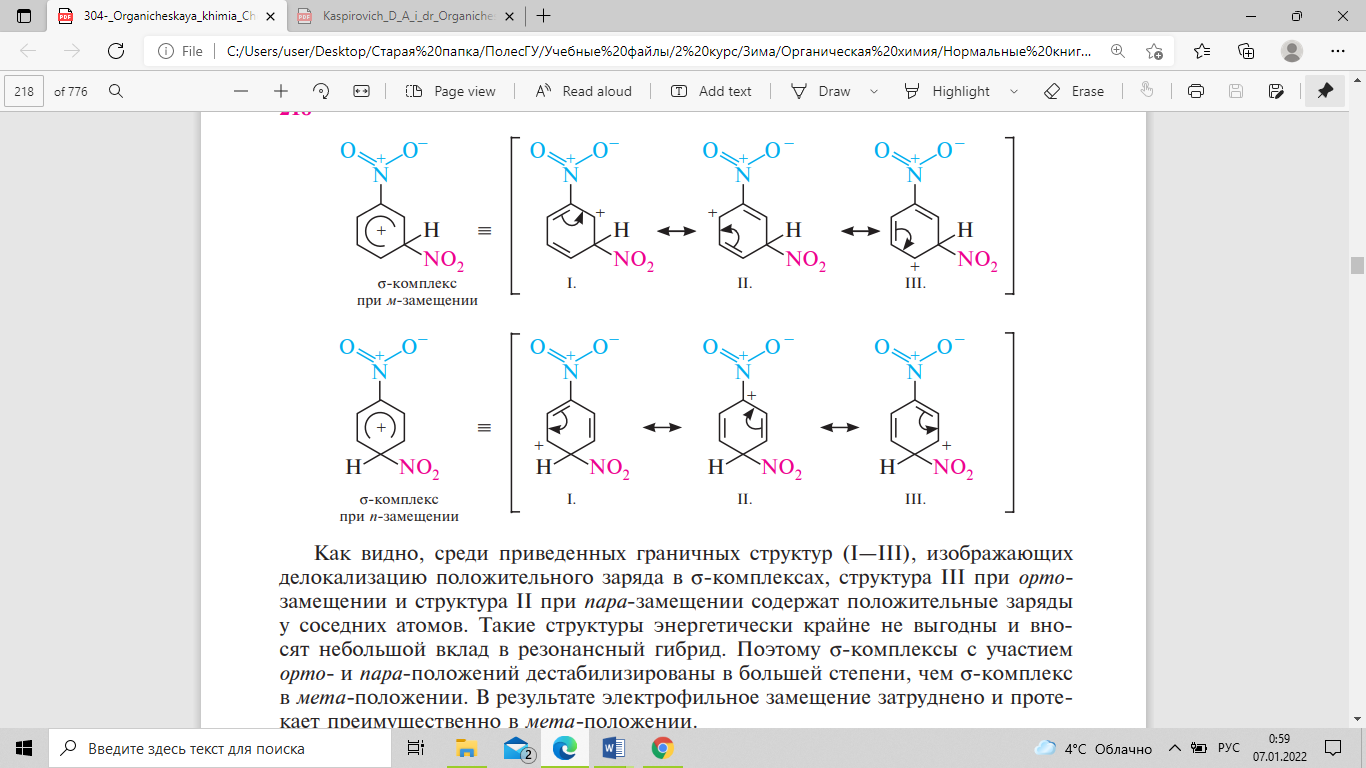 Такие
структуры энергетически крайне невыгодны
и вносят небольшой вклад в резонансный
гибрид. Поэтому σ-комплексы с участием
орто- и пара-положений дестабилизированы
в большей степени, чем σ-комплекс в
мета-положении. В результате электрофильное
замещение затруднено и протекает
преимущественно в мета-положении. В
большинстве случаев статический и
динамический факторы действуют
согласованно. Но если их влияние
проявляется в противоположных
направлениях, то решающее значение на
направление электрофильного замещения
оказывает динамический фактор. Наглядным
примером могут служить арилгалогениды,
в которых атомы галогена проявляют
электроноакцепторные свойства, но
вместе с тем направляют электрофильное
замещение в орто- и пара-положения.
Причиной такого поведения галогенов в
качестве заместителя является
несогласованное действие статического
и динамического факторов. Как известно,
атом галогена в бензольном ядре проявляет
отрицательный индуктивный и положительный
мезомерный эффекты, причем в статическом
состоянии –I-эффект значительнее
+М-эффекта. В результате происходит
смещение электронной плотности
бензольного кольца в сторону атома
галогена и, следовательно, реакционная
способность цикла по отношению к
электрофильным реагентам снижается.
Следовательно, в статическом состоянии
галогены, подобно ориентантам II рода,
затрудняют электрофильное замещение.
Однако в процессе реакции неподеленные
пары электронов атома галогена, которые
находятся в сопряжении с π-электронной
системой бензольного кольца, принимают
участие в дополнительной стабилизации
σ-комплексов, образующихся при орто- и
пара-замещении, но не участвуют в
стабилизации мета-σ-комплекса. Поэтому
галогены выступают как заместители I
рода и направляют электрофильное
замещение в орто- и пара-положения. Кроме
заместителей I и II рода, имеется небольшое
число заместителей, проявляющих смешанное
действие (—CH2NO2, —CH2Hal, —CH2OH, —CHHal2 и др.).
Эти заместители несколько затрудняют
электрофильное замещение в бензольном
ядре, но в результате реакции, как
правило, образуется смесь примерно
равных количеств орто-, мета- и
пара-изомеров.
Такие
структуры энергетически крайне невыгодны
и вносят небольшой вклад в резонансный
гибрид. Поэтому σ-комплексы с участием
орто- и пара-положений дестабилизированы
в большей степени, чем σ-комплекс в
мета-положении. В результате электрофильное
замещение затруднено и протекает
преимущественно в мета-положении. В
большинстве случаев статический и
динамический факторы действуют
согласованно. Но если их влияние
проявляется в противоположных
направлениях, то решающее значение на
направление электрофильного замещения
оказывает динамический фактор. Наглядным
примером могут служить арилгалогениды,
в которых атомы галогена проявляют
электроноакцепторные свойства, но
вместе с тем направляют электрофильное
замещение в орто- и пара-положения.
Причиной такого поведения галогенов в
качестве заместителя является
несогласованное действие статического
и динамического факторов. Как известно,
атом галогена в бензольном ядре проявляет
отрицательный индуктивный и положительный
мезомерный эффекты, причем в статическом
состоянии –I-эффект значительнее
+М-эффекта. В результате происходит
смещение электронной плотности
бензольного кольца в сторону атома
галогена и, следовательно, реакционная
способность цикла по отношению к
электрофильным реагентам снижается.
Следовательно, в статическом состоянии
галогены, подобно ориентантам II рода,
затрудняют электрофильное замещение.
Однако в процессе реакции неподеленные
пары электронов атома галогена, которые
находятся в сопряжении с π-электронной
системой бензольного кольца, принимают
участие в дополнительной стабилизации
σ-комплексов, образующихся при орто- и
пара-замещении, но не участвуют в
стабилизации мета-σ-комплекса. Поэтому
галогены выступают как заместители I
рода и направляют электрофильное
замещение в орто- и пара-положения. Кроме
заместителей I и II рода, имеется небольшое
число заместителей, проявляющих смешанное
действие (—CH2NO2, —CH2Hal, —CH2OH, —CHHal2 и др.).
Эти заместители несколько затрудняют
электрофильное замещение в бензольном
ядре, но в результате реакции, как
правило, образуется смесь примерно
равных количеств орто-, мета- и
пара-изомеров.