

36. Реакции радикального замещения и окисления в боковой цепи. Причины устойчивости бензильных радикалов.
Ответ.
Алкилбензолы, в отличие от незамещенного
бензола, окисляются
значительно легче. При действии сильных
окислителей (KMnO4, K2Cr2O7 и др.) окислению
подвергаются боковые углеродные цепи.
Продуктами окисления являются
ароматические карбоновые кислоты.
Причем каждый алкильный радикал в
бензольном кольце, независимо от длины
углеродной цепи, окисляется в карбоксильную
группу.
 Если
в бензольном кольце имеется несколько
заместителей, то путем подбора оптимальных
условий можно провести их последовательное
окисление. Алкильные группы по реакционной
способности к действию окислителей
располагаются в следующей последовательности:
—CHR2 > —CH2R > —CH3. Третичные алкильные
группы у бензольного ядра до карбоксильной
группы не окисляются. Окисление
алкилбензолов является важным способом
получения ароматических карбоновых
кислот. Взаимодействие гомологов бензола
с галогенами (хлором или бромом) в
условиях свободнорадикального
замещения осуществляется с участием
боковой
цепи. При этом на атом галогена замещается,
как правило, атом водорода при атоме
углерода, непосредственно связанном с
бензольным кольцом (α-положение):
Если
в бензольном кольце имеется несколько
заместителей, то путем подбора оптимальных
условий можно провести их последовательное
окисление. Алкильные группы по реакционной
способности к действию окислителей
располагаются в следующей последовательности:
—CHR2 > —CH2R > —CH3. Третичные алкильные
группы у бензольного ядра до карбоксильной
группы не окисляются. Окисление
алкилбензолов является важным способом
получения ароматических карбоновых
кислот. Взаимодействие гомологов бензола
с галогенами (хлором или бромом) в
условиях свободнорадикального
замещения осуществляется с участием
боковой
цепи. При этом на атом галогена замещается,
как правило, атом водорода при атоме
углерода, непосредственно связанном с
бензольным кольцом (α-положение):
 Такое
направление замещения обусловлено
образованием в качестве промежуточной
активной частицы свободного радикала
бензильного типа, в котором электронная
плотность значительно делокализована
за счет сопряжения с бензольным кольцом:
Такое
направление замещения обусловлено
образованием в качестве промежуточной
активной частицы свободного радикала
бензильного типа, в котором электронная
плотность значительно делокализована
за счет сопряжения с бензольным кольцом:
37. Классификация, номенклатура, изомерия галогенуглеводородов.
Ответ.
Галогенопроизводными углеводородов
называют продукты замещения в углеводородах
одного или нескольких атомов водорода
атомами галогенов. Галогенопроизводные
углеводородов в зависимости от природы
углеводородного радикала подразделяют
на алифатические, алициклические и
ароматические. В ряду алифатических
галогенопроизводных углеводородов
различают насыщенные (галогеналканы)
и ненасыщенные (галогеналкены,
галогеналкины). Ароматические
галогенопроизводные углеводородов
делят на соединения, в которых атом
галогена непосредственно связан с
ароматическим ядром (галогенарены), и
вещества, содержащие атом галогена в
боковой цепи (арилалкилгалогениды). В
соответствии с природой атома галогена
галогенопроизводные углеводородов
подразделяют на фтор-, хлор-, бром-,
йодпроизводные. По числу атомов галогена
в молекуле различают моно-, ди-, три- и
полигалогенопроизводные углеводородов.
По заместительной номенклатуре
IUPAC названия галогенопроизводных
углеводородов составляют аналогично
названиям соответствующих углеводородов.
Входящие в их состав атомы галогенов
обозначают в названии в виде префикса,
к которому прибавляют название
родоначальной структуры. За родоначальную
структуру в алифатических галогенопроизводных
углеводородов принимается главная
углеродная цепь, в алициклических и
ароматических — цикл. Если при
родоначальной структуре имеется
несколько заместителей, которыми, кроме
атомов галогенов, могут быть и
углеводородные радикалы, то в названии
их перечисляют в алфавитном порядке.
Атомы углерода родоначальной структуры
нумеруют в данном случае таким образом,
чтобы заместитель, который обозначен
в названии первым, получил возможно
меньший номер. В галогеналкенах и
галогеналкинах нумерацию атомов углерода
главной цепи проводят так, чтобы возможно
меньшие номера получили атомы углерода
кратной связи. Для простейших
галогенопроизводных углеводородов
широко используют радикало-функциональную
номенклатуру, согласно которой название
составляют из названия углеводородного
радикала, связанного с атомом галогена
и суффикса -фторид, -хлорид, -бромид или
–йодид. Полностью галогенированные
углеводороды (все атомы водорода замещены
атомами галогена) называют
пергалогенированными (в названиях
используют префикс пер-). Для некоторых
галогеноуглеводородов приняты тривиальные
названия:
 Для
галогенопроизводных углеводородов
характерны структурная, геометрическая
и оптическая изомерии. Структурная
изомерия обусловлена разной структурой
углеродного скелета молекулы и разным
положением атомов галогенов в цепи.
Дигалогенопроизводные могут быть
представлены несколькими структурными
изомерами, отличающимися взаимным
расположением атомов галогенов.
Дигалогенопроизводные углеводородов
с атомами галогенов у одного и того же
атома углерода называют геминальными
(сокращенно гем), у соседних атомов
углерода — вицинальными (сокр. виц).
Для
галогенопроизводных углеводородов
характерны структурная, геометрическая
и оптическая изомерии. Структурная
изомерия обусловлена разной структурой
углеродного скелета молекулы и разным
положением атомов галогенов в цепи.
Дигалогенопроизводные могут быть
представлены несколькими структурными
изомерами, отличающимися взаимным
расположением атомов галогенов.
Дигалогенопроизводные углеводородов
с атомами галогенов у одного и того же
атома углерода называют геминальными
(сокращенно гем), у соседних атомов
углерода — вицинальными (сокр. виц).
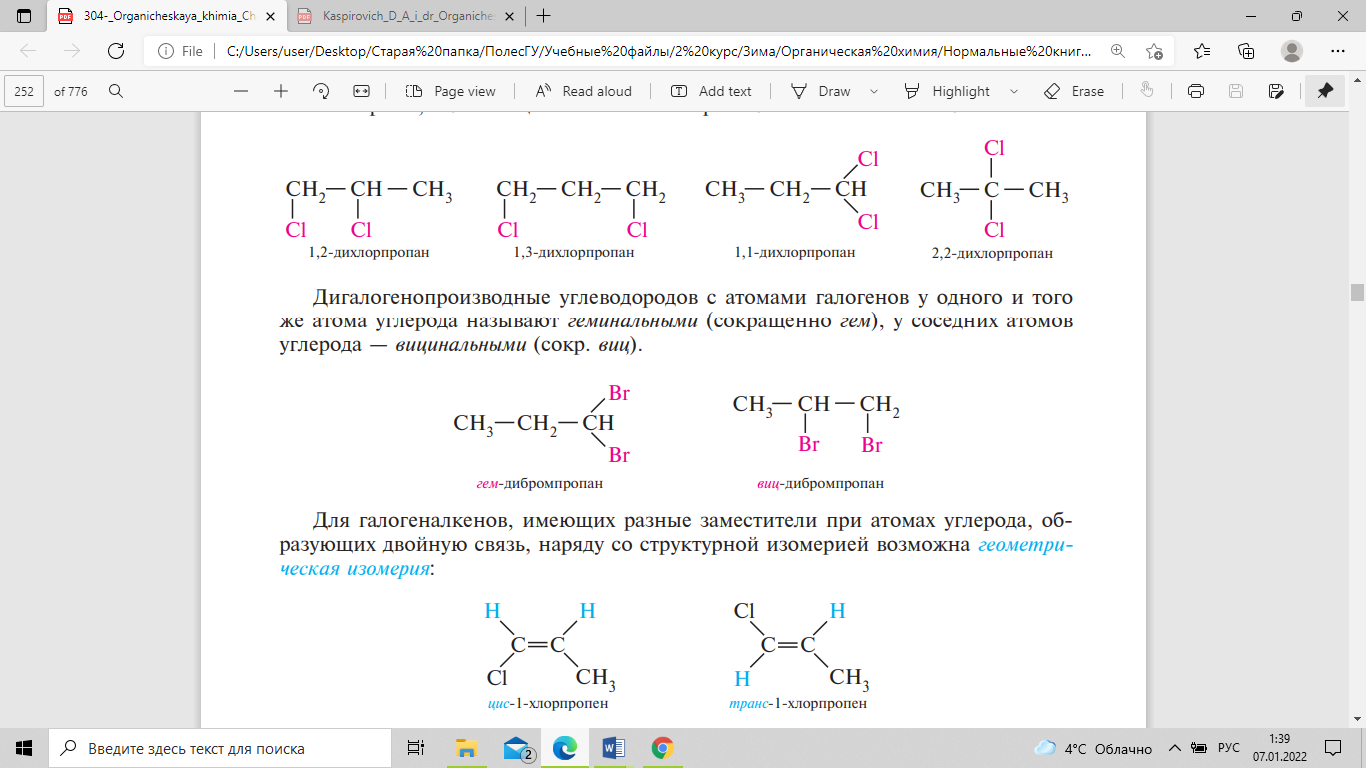 Для
галогеналкенов, имеющих разные заместители
при атомах углерода, образующих двойную
связь, наряду со структурной изомерией
возможна геометрическая изомерия:
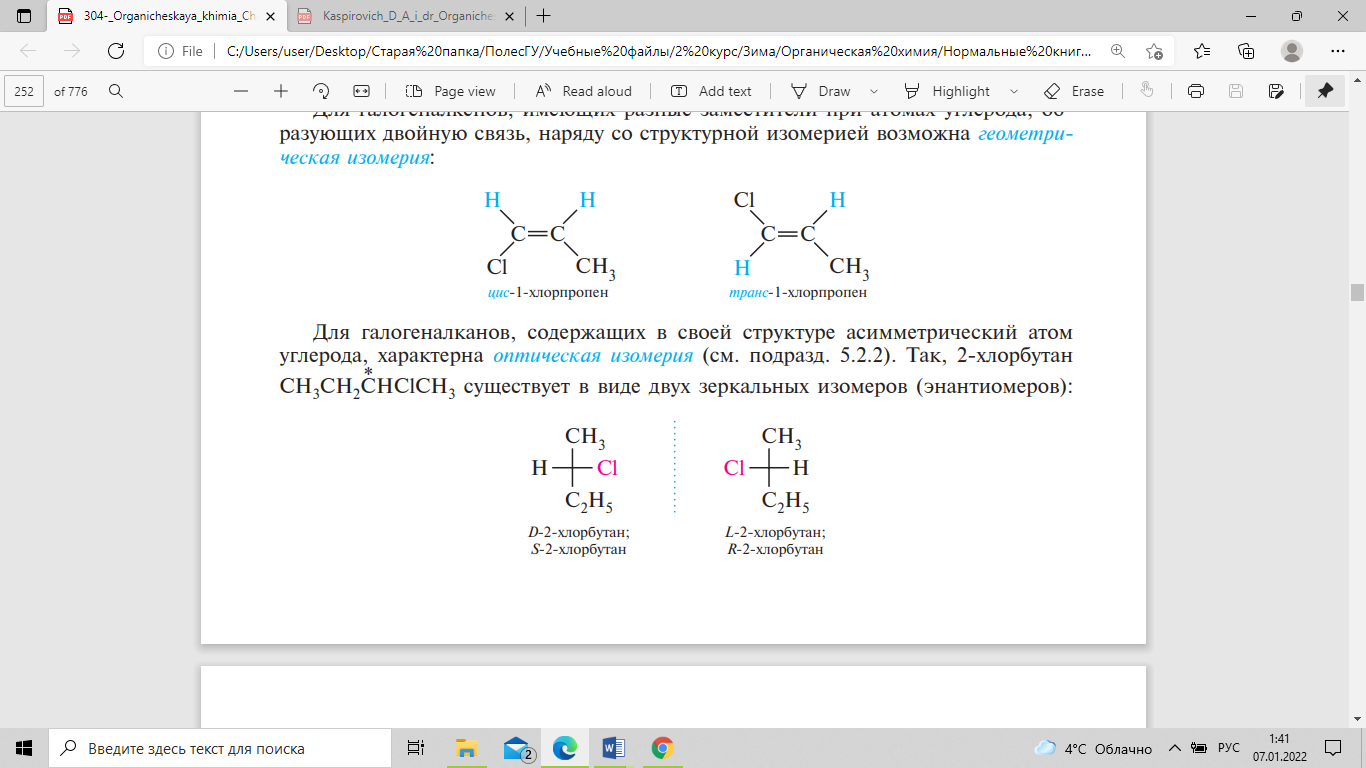
Для
галогеналканов, содержащих в своей
структуре асимметрический атом углерода,
характерна оптическая изомерия. Так,
2-хлорбутан существует в виде двух
зеркальных изомеров (энантиомеров):
Для
галогеналкенов, имеющих разные заместители
при атомах углерода, образующих двойную
связь, наряду со структурной изомерией
возможна геометрическая изомерия:
Для
галогеналканов, содержащих в своей
структуре асимметрический атом углерода,
характерна оптическая изомерия. Так,
2-хлорбутан существует в виде двух
зеркальных изомеров (энантиомеров):
38. Реакции нуклеофильного замещения атома галогена, их использование в синтезе органических соединений различных классов (спиртов, простых и сложных эфиров, аминов, тиолов и сульфидов, нитроалканов, нитрилов). Представление об идеализированных механизмах SN1 и SN2.
Ответ.
Галогеналканы являются весьма
реакционноспособными веществами.
Наиболее характерны для них реакции
нуклеофильного замещения (SN ) и отщепления
(E ). Они также способны к взаимодействию
с металлами и восстановлению. Галогеналканы
являются электрофильными реагентами.
Их электрофильные свойства обусловлены
полярностью связи C—Hal. Поскольку атом
галогена проявляет большую
электроотрицательность, чем атом
углерода, электронная плотность связи
C–Hal в галогеналканах смещена к атому
галогена. В результате атом галогена
приобретает частичный δ–, а атом углерода
— частичный δ+ заряды. Электронодефицитный
атом углерода становится электрофильным
центром молекулы галогеналкана и может
быть атакован нуклеофильным реагентом.
В процессе атаки нуклеофил предоставляет
пару электронов для образования
химической связи с электронодефицитным
атомом С, а атом галогена отщепляется
от молекулы галогеналкана с электронной
парой связи C—Hal. Такую реакцию называют
реакцией нуклеофильного замещения и
обозначают символом SN. Нуклеофильными
реагентами могут быть вещества, содержащие
в молекулах атомы с неподеленными парами
электронов (NH3, R—NH2, R2NH, HOH и др.), либо
вещества, образующие при диссоциации
анионы (нуклеофильные частицы): NaOH (OH–),
C2H5ONa (C2H5O–), KCN (CN–), NaSH (SH–),
NaNO2
(NO2–),
CH3COONa
(CH3COO–),
KBr
(Br–),
KI
(I–)
и др. Реакционная способность галогеналканов
в реакциях SN уменьшается в ряду: R—I >
R—Br > R—Cl >> R—F. В зависимости от
строения галогеналкана, природы
нуклеофила и растворителя реакции
нуклеофильного замещения протекают по
двум основным механизмам: механизм SN2
и механизм SN1.
По механизму SN2
реакция происходит в одну стадию через
образование переходного состояния, в
построении которого принимает участие
как молекула галогеналкана, так и
нуклеофильный реагент. В механизме SN2
нуклеофил атакует электрофильный центр
молекулы галогеналкана (атом углерода,
связанный с атомом галогена) со стороны,
противоположной связи C—Hal (атака с
тыла). При сближении нуклеофила с
галогеналканом в их молекулах происходит
перераспределение электронной плотности
химических связей. В результате образуется
переходное состояние, представляющее
собой предельно неустойчивое сочетание
двух реагентов, в котором связь C—Hal
ослабляется и начинает формироваться
связь C—Nu. Переходное состояние находится
в равновесии с исходными реагентами.
По мере дальнейшего сближения реагентов
в переходном состоянии происходит
синхронный процесс разрыва связи C—Hal
и образование связи C—Nu:
 Буква
S указывает на замещение, N — на
нуклеофильный тип реакции, а цифра 2
обозначает, что реакция является
бимолекулярной, то есть в стадии,
определяющей скорость реакции в целом
(в данном случае образование переходного
состояния), участвуют два реагента
(галогеналкан и нуклеофил). По механизму
SN1
реакция протекает в две стадии:
Буква
S указывает на замещение, N — на
нуклеофильный тип реакции, а цифра 2
обозначает, что реакция является
бимолекулярной, то есть в стадии,
определяющей скорость реакции в целом
(в данном случае образование переходного
состояния), участвуют два реагента
(галогеналкан и нуклеофил). По механизму
SN1
реакция протекает в две стадии:
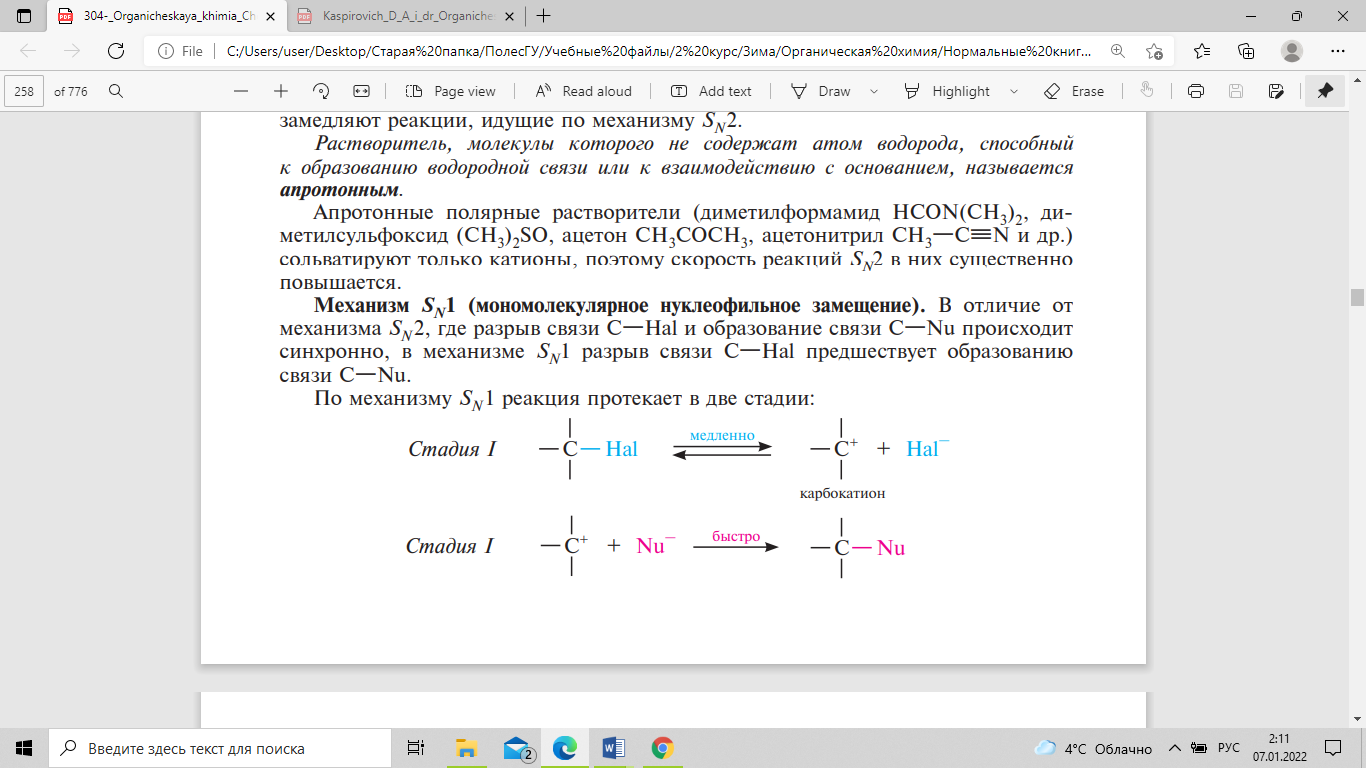 На
стадии I происходит ионизация молекулы
галогеналкана с образованием карбокатиона
и галогенид-иона. Процесс ионизации
протекает медленно, а поэтому он
определяет скорость всей реакции. В
ионизации галогеналкана оказывает
содействие растворитель. На стадии II
образовавшийся катион быстро
взаимодействует с нуклеофильным
реагентом, образуя конечный продукт
реакции. Механизм называют мономолекулярным,
так как на стадии, определяющей скорость
всего процесса (стадия I) принимает
участие молекула только одного реагента
— галогеналкана. Такой механизм
обозначают SN1. Первичные галогеналканы
обычно реагируют по механизму SN2,
третичные — по SN1. Вторичные галогеналканы,
в зависимости от природы нуклеофила и
растворителя, могут реагировать как по
механизму SN2, так и по механизму SN1.
Галогеналканы при гидролизе
образуют спирты.
Реакция с водой протекает медленно и
является обратимой, поэтому обычно
гидролиз проводят в присутствии водных
растворов щелочей или карбонатов
щелочных металлов:
На
стадии I происходит ионизация молекулы
галогеналкана с образованием карбокатиона
и галогенид-иона. Процесс ионизации
протекает медленно, а поэтому он
определяет скорость всей реакции. В
ионизации галогеналкана оказывает
содействие растворитель. На стадии II
образовавшийся катион быстро
взаимодействует с нуклеофильным
реагентом, образуя конечный продукт
реакции. Механизм называют мономолекулярным,
так как на стадии, определяющей скорость
всего процесса (стадия I) принимает
участие молекула только одного реагента
— галогеналкана. Такой механизм
обозначают SN1. Первичные галогеналканы
обычно реагируют по механизму SN2,
третичные — по SN1. Вторичные галогеналканы,
в зависимости от природы нуклеофила и
растворителя, могут реагировать как по
механизму SN2, так и по механизму SN1.
Галогеналканы при гидролизе
образуют спирты.
Реакция с водой протекает медленно и
является обратимой, поэтому обычно
гидролиз проводят в присутствии водных
растворов щелочей или карбонатов
щелочных металлов:
![]() При
действии на галогеналканы алкоголятов
и фенолятов
образуются простые
эфиры.
Третичные галогеналканы образуют в
качестве побочных продуктов алкены:
При
действии на галогеналканы алкоголятов
и фенолятов
образуются простые
эфиры.
Третичные галогеналканы образуют в
качестве побочных продуктов алкены:
 Реакция
открыта в 1851 году Вильямсоном и
используется в качестве одного из лучших
методов получения простых эфиров. При
действии на галогеналканы солей
карбоновых
кислот
в среде апротонного полярного растворителя
(диметилформамид, диметилсульфоксид)
с высокими выходами образуются сложные
эфиры (по механизму SN2):
Реакция
открыта в 1851 году Вильямсоном и
используется в качестве одного из лучших
методов получения простых эфиров. При
действии на галогеналканы солей
карбоновых
кислот
в среде апротонного полярного растворителя
(диметилформамид, диметилсульфоксид)
с высокими выходами образуются сложные
эфиры (по механизму SN2):
![]() При
взаимодействии галогеналканов с избытком
аммиака
образуется смесь первичных, вторичных
и третичных аминов, а также соли
четвертичных аммониевых оснований.
При
взаимодействии галогеналканов с избытком
аммиака
образуется смесь первичных, вторичных
и третичных аминов, а также соли
четвертичных аммониевых оснований.
![]() Аналогично реагируют галогеналканы с
алкил- и ариламинами. Поскольку цианид-ион
является амбидентным нуклеофилом, при
взаимодействии галогеналканов с солями
циановодородной кислоты образуются
нитрилы
или изонитрилы
(изоцианиды) в зависимости от условий
проведения реакции. Первичные и вторичные
галогеналканы с солями щелочных металлов
циановодородной кислоты (KCN, NaCN) в среде
апротонного полярного растворителя с
хорошими выходами образуют нитрилы
(механизм SN2):
Аналогично реагируют галогеналканы с
алкил- и ариламинами. Поскольку цианид-ион
является амбидентным нуклеофилом, при
взаимодействии галогеналканов с солями
циановодородной кислоты образуются
нитрилы
или изонитрилы
(изоцианиды) в зависимости от условий
проведения реакции. Первичные и вторичные
галогеналканы с солями щелочных металлов
циановодородной кислоты (KCN, NaCN) в среде
апротонного полярного растворителя с
хорошими выходами образуют нитрилы
(механизм SN2):
![]() Основными
продуктами реакции вторичных и третичных
галогеналканов с серебра цианидом в
среде протонного полярного растворителя
являются изонитрилы (изоцианиды)
(механизм SN1).
Взаимодействие
галогеналканов с солями
азотистой
кислоты, содержащими амбидентный
нитрит-ион, протекает в зависимости от
условий проведения реакции с образованием
нитросоединений
или эфиров
азотистой
кислоты. Первичные и вторичные
галогеналканы с натрия нитритом в
условиях реакции SN2 образуют преимущественно
нитросоединения:
Основными
продуктами реакции вторичных и третичных
галогеналканов с серебра цианидом в
среде протонного полярного растворителя
являются изонитрилы (изоцианиды)
(механизм SN1).
Взаимодействие
галогеналканов с солями
азотистой
кислоты, содержащими амбидентный
нитрит-ион, протекает в зависимости от
условий проведения реакции с образованием
нитросоединений
или эфиров
азотистой
кислоты. Первичные и вторичные
галогеналканы с натрия нитритом в
условиях реакции SN2 образуют преимущественно
нитросоединения:
![]() Вторичные
и третичные галогеналканы с серебра
нитритом AgNO2 в условиях реакции SN1
образуют с хорошими выходами эфиры
азотистой кислоты:
Реакция
Финкельштейна позволяет заменить в
молекуле галогеналкана один атом
галогена другим. Взаимодействие галогенов
с солями галогеноводородных кислот
является обратимым процессом. Для
смещения равновесия вправо используют
разную растворимость исходных веществ
и продуктов реакции. Реакция имеет
практическое значение для получения
первичных фтор- и йодалканов из более
доступных хлор- и бромпроизводных. Для
получения йодидов реакцию проводят в
ацетоне, так как натрия йодид растворим
в ацетоне, а образующиеся в процессе
взаимодействия NaCl или NaBr выпадают в
осадок:
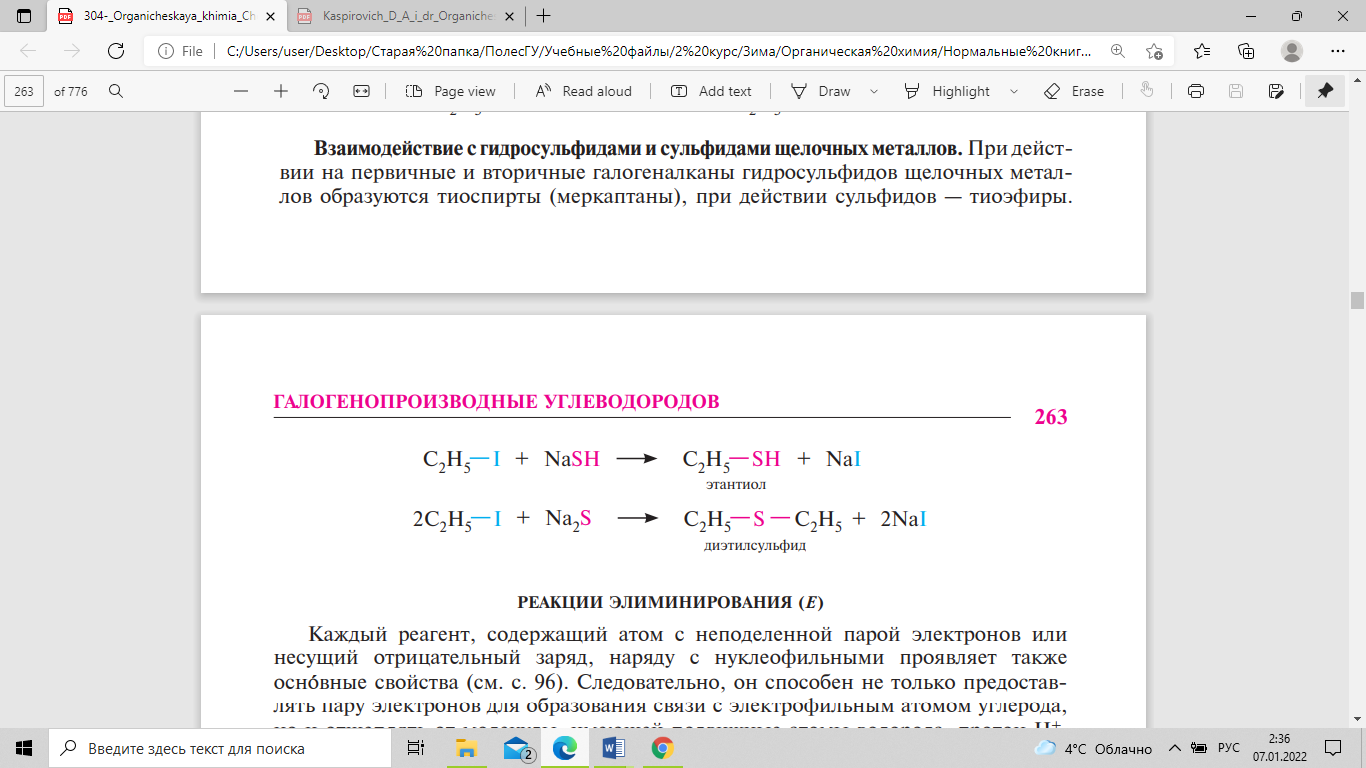
При
действии на первичные и вторичные
галогеналканы гидросульфидов
щелочных
металлов
образуются тиоспирты
(меркаптаны), при действии сульфидов
— тиоэфиры.
Вторичные
и третичные галогеналканы с серебра
нитритом AgNO2 в условиях реакции SN1
образуют с хорошими выходами эфиры
азотистой кислоты:
Реакция
Финкельштейна позволяет заменить в
молекуле галогеналкана один атом
галогена другим. Взаимодействие галогенов
с солями галогеноводородных кислот
является обратимым процессом. Для
смещения равновесия вправо используют
разную растворимость исходных веществ
и продуктов реакции. Реакция имеет
практическое значение для получения
первичных фтор- и йодалканов из более
доступных хлор- и бромпроизводных. Для
получения йодидов реакцию проводят в
ацетоне, так как натрия йодид растворим
в ацетоне, а образующиеся в процессе
взаимодействия NaCl или NaBr выпадают в
осадок:
При
действии на первичные и вторичные
галогеналканы гидросульфидов
щелочных
металлов
образуются тиоспирты
(меркаптаны), при действии сульфидов
— тиоэфиры.
39. Соединения с повышенной подвижностью атома галогена. (аллил- и бензилгалогениды). Соединения с пониженной подвижностью атома галогена (винилхлорид и хлорбензол). Реакции элиминирования галогеноводорода. Правило Зайцева.
Ответ.
Аллил-
и бензолгалогениды
очень легко вступают в реакции
нуклеофиьного замещения. Гидролиз
аллил- и бензилбромидов осуществляется
кипячением с водой, а при использовании
вместо воды (слабый нуклеофил) ––
водного раствора гидраксида натрия
(сильный нуклеофил) – реакция происходит
при комнатной температуре:
 Каждый
из рассматриваемых субстратов образует
катион аллильного типа. Аллильный
карбкатион стабилизирован вследствие
сопряжения вакантной р-орбитали с
соседней π-связью и делокализации заряда
по сопряженной системе. Для каждого из
исходных субстратов карбкатион аллильного
типа может быть представлен граничными
каноническими структурами с различным
распределением положительного заряда.
Взаимодействие аллильного карбкатиона
на второй стадии с нуклеофилом (водой)
приводит к образованию смеси спиртов.
Реакционная способность аллилгалогенидов
в реакциях нуклеофильного замещения
обусловлена стабильностью интермедиата
(алильного карбкатиона), что уменьшает
энергию активации, необходимую для его
образования. Бензилгалогениды аналогично
аллилгалогенидам легко вступают в
реакции мономолекулярного нуклеофильного
замещения. Во многих случаях и кинетика,
и стереохимия реакций бензилгалогенидов
соответствует мономолекулярному
механизму. Образующийся в результате
диссоциации субстрата на медленной
стадии бензильный карбкатион стабилизирован
вследствие делокализации положительного
заряда по сопряженной системе
ароматического кольца. Заместители,
которые стабилизируют интермедиат,
будут повышать реакционную способность
бензилгалогенидов. Так, скорость
взаимодействия 4-метоксибензилхлорида
с этанолом выше, чем незамещенного
бензилхлорида, что объясняется
дополнительным влиянием метоксигруппы
на стабилизацию карбкатиона. Нуклеофильное
замещение в ряду аллил- и бензилгалогенидов
может протекать и по механизму
бимолекулярного нуклеофильного замещения
SN2, если для этого созданы соответствующие
условия: высокая концентрация нуклеофила;
малополярная слабоионизирующая среда).
В молекулах аллилгалогенидов, в отличие
от винилгалогенидов, атом галогена
обладает повышенной подвижностью.
Аллилгалогениды вступают в реакции
нуклеофильного замещения легче, чем
галогеналканы. Замещение, как правило,
происходит по механизму SN1. Винилхлорид
- бесцветный газ со слабым сладковатым
запахом, имеющий формулу C2H3Cl и
представляющий собой простейшее
хлорпроизводное этилена. В молекуле
винилхлорида связь C-Cl более короткая
и более прочная, чем аналогичная связь
в молекуле хлорэтана. Укорочение длины
связи обусловлено p,π-сопряжением
π-орбиталей кратной связи с неподелённой
электронной парой атома хлора и
образованием единой делокализованной
системы π-электронов. Химические свойства
винилхлорида определяются как наличием
двойной связи, так и атома хлора. Подобно
алкенам, винилхлорид вступает в реакции
присоединения по кратной связи. В отличие
от галогеналкенов винилгалогениды
обладают низкой реакционной способностью
в обычных реакциях нуклеофильного
замещения и элиминирования. Винилгалогениды
не удается превратить при действии
обычных нуклеофильных реагентов в
спирт, простые и сложные эфиры, амины и
т.д. Под действием оснований, особенно
при нагревании, вместо замещения
протекают реакции полимеризации. В
отсутствии кислорода и света при обычных
условиях чистый винилхлорид может
существовать достаточно долго, не
претерпевая каких-либо изменений; однако
появление свободных радикалов, вызываемое
как фотохимически, так и термохимически,
приводит к его быстрой полимеризации.
Хлорбензол
— ароматическое органическое соединение,
имеющее формулу C6H5Cl, бесцветная горючая
жидкость с характерным запахом. Хлорбензол
является важным органическим растворителем,
кроме того он применяется в органическом
синтезе, например он применяется в
синтезе пестицидов (например, ДДТ может
быть получен реакцией его с хлоралем
(трихлорацетальдегидом)). Также применяется
в производстве фенола. Высокая активность
аллилгалогенидов в реакциях нуклеофильного
замещения объясняется их склонностью
к ионизации, поскольку при этом образуется
весьма устойчивый аллильный катион,
обусловленная делокализацией
положительного заряда по сопряженной
системе. Каждый
реагент, содержащий атом с неподеленной
парой электронов или несущий отрицательный
заряд, наряду с нуклеофильными проявляет
также основные свойства. Следовательно,
он способен не только предоставлять
пару электронов для образования связи
с электрофильным атомом углерода, но и
отщеплять от молекулы, имеющей подвижные
атомы водорода, протон Н+. В молекуле
галогеналкана приобретают подвижность
атомы водорода у β-углеродного атома
вследствие –I-эффекта атома галогена,
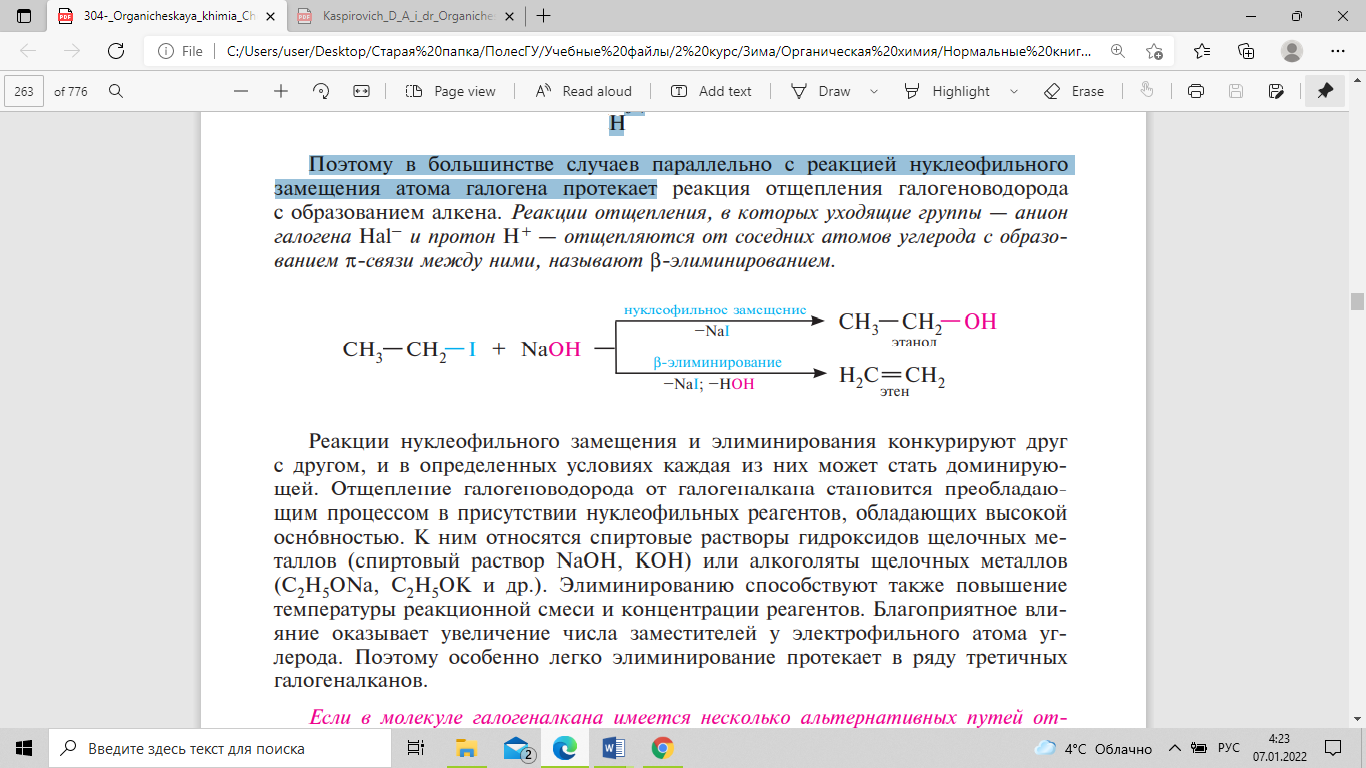
поэтому в большинстве случаев параллельно
с реакцией нуклеофильного замещения
атома галогена протекает реакция
отщепления галогеноводорода с образованием
алкена. Реакции отщепления, в которых
уходящие группы — анион Нal– и протон
Н+ — отщепляются от соседних атомов
углерода с образованием π-связи между
ними, называют β-элиминированием.
Каждый
из рассматриваемых субстратов образует
катион аллильного типа. Аллильный
карбкатион стабилизирован вследствие
сопряжения вакантной р-орбитали с
соседней π-связью и делокализации заряда
по сопряженной системе. Для каждого из
исходных субстратов карбкатион аллильного
типа может быть представлен граничными
каноническими структурами с различным
распределением положительного заряда.
Взаимодействие аллильного карбкатиона
на второй стадии с нуклеофилом (водой)
приводит к образованию смеси спиртов.
Реакционная способность аллилгалогенидов
в реакциях нуклеофильного замещения
обусловлена стабильностью интермедиата
(алильного карбкатиона), что уменьшает
энергию активации, необходимую для его
образования. Бензилгалогениды аналогично
аллилгалогенидам легко вступают в
реакции мономолекулярного нуклеофильного
замещения. Во многих случаях и кинетика,
и стереохимия реакций бензилгалогенидов
соответствует мономолекулярному
механизму. Образующийся в результате
диссоциации субстрата на медленной
стадии бензильный карбкатион стабилизирован
вследствие делокализации положительного
заряда по сопряженной системе
ароматического кольца. Заместители,
которые стабилизируют интермедиат,
будут повышать реакционную способность
бензилгалогенидов. Так, скорость
взаимодействия 4-метоксибензилхлорида
с этанолом выше, чем незамещенного
бензилхлорида, что объясняется
дополнительным влиянием метоксигруппы
на стабилизацию карбкатиона. Нуклеофильное
замещение в ряду аллил- и бензилгалогенидов
может протекать и по механизму
бимолекулярного нуклеофильного замещения
SN2, если для этого созданы соответствующие
условия: высокая концентрация нуклеофила;
малополярная слабоионизирующая среда).
В молекулах аллилгалогенидов, в отличие
от винилгалогенидов, атом галогена
обладает повышенной подвижностью.
Аллилгалогениды вступают в реакции
нуклеофильного замещения легче, чем
галогеналканы. Замещение, как правило,
происходит по механизму SN1. Винилхлорид
- бесцветный газ со слабым сладковатым
запахом, имеющий формулу C2H3Cl и
представляющий собой простейшее
хлорпроизводное этилена. В молекуле
винилхлорида связь C-Cl более короткая
и более прочная, чем аналогичная связь
в молекуле хлорэтана. Укорочение длины
связи обусловлено p,π-сопряжением
π-орбиталей кратной связи с неподелённой
электронной парой атома хлора и
образованием единой делокализованной
системы π-электронов. Химические свойства
винилхлорида определяются как наличием
двойной связи, так и атома хлора. Подобно
алкенам, винилхлорид вступает в реакции
присоединения по кратной связи. В отличие
от галогеналкенов винилгалогениды
обладают низкой реакционной способностью
в обычных реакциях нуклеофильного
замещения и элиминирования. Винилгалогениды
не удается превратить при действии
обычных нуклеофильных реагентов в
спирт, простые и сложные эфиры, амины и
т.д. Под действием оснований, особенно
при нагревании, вместо замещения
протекают реакции полимеризации. В
отсутствии кислорода и света при обычных
условиях чистый винилхлорид может
существовать достаточно долго, не
претерпевая каких-либо изменений; однако
появление свободных радикалов, вызываемое
как фотохимически, так и термохимически,
приводит к его быстрой полимеризации.
Хлорбензол
— ароматическое органическое соединение,
имеющее формулу C6H5Cl, бесцветная горючая
жидкость с характерным запахом. Хлорбензол
является важным органическим растворителем,
кроме того он применяется в органическом
синтезе, например он применяется в
синтезе пестицидов (например, ДДТ может
быть получен реакцией его с хлоралем
(трихлорацетальдегидом)). Также применяется
в производстве фенола. Высокая активность
аллилгалогенидов в реакциях нуклеофильного
замещения объясняется их склонностью
к ионизации, поскольку при этом образуется
весьма устойчивый аллильный катион,
обусловленная делокализацией
положительного заряда по сопряженной
системе. Каждый
реагент, содержащий атом с неподеленной
парой электронов или несущий отрицательный
заряд, наряду с нуклеофильными проявляет
также основные свойства. Следовательно,
он способен не только предоставлять
пару электронов для образования связи
с электрофильным атомом углерода, но и
отщеплять от молекулы, имеющей подвижные
атомы водорода, протон Н+. В молекуле
галогеналкана приобретают подвижность
атомы водорода у β-углеродного атома
вследствие –I-эффекта атома галогена,
поэтому в большинстве случаев параллельно
с реакцией нуклеофильного замещения
атома галогена протекает реакция
отщепления галогеноводорода с образованием
алкена. Реакции отщепления, в которых
уходящие группы — анион Нal– и протон
Н+ — отщепляются от соседних атомов
углерода с образованием π-связи между
ними, называют β-элиминированием.
 Реакции
конкурируют друг с другом, и в определенных
условиях каждая из них может стать
доминирующей. Отщепление галогеноводорода
от галогеналкана становится преобладающим
процессом в присутствии нуклеофильных
реагентов, обладающих высокой основностью.
К ним относятся спиртовые растворы
гидроксидов щелочных металлов (спиртовый
раствор NaOH, KOH) или алкоголяты щелочных
металлов (C2H5ONa, С2Н5ОК и др.). Элиминированию
способствуют также повышение температуры
реакционной смеси и концентрации
реагентов. Благоприятное влияние
оказывает увеличение числа заместителей
у электрофильного атома углерода.
Поэтому особенно легко элиминирование
протекает в ряду третичных галогеналканов.
Если в молекуле галогеналкана имеется
несколько альтернативных путей отщепления
галогеноводорода, то преимущественно
реализуется из них тот, при котором
двойная связь образуется у наиболее
замещенного атома углерода; то есть
вместе с галогеном уходит водород от
наименее гидрогенизированного соседнего
атома углерода. Эта закономерность
получила название «правило
Зайцева»:
Реакции
конкурируют друг с другом, и в определенных
условиях каждая из них может стать
доминирующей. Отщепление галогеноводорода
от галогеналкана становится преобладающим
процессом в присутствии нуклеофильных
реагентов, обладающих высокой основностью.
К ним относятся спиртовые растворы
гидроксидов щелочных металлов (спиртовый
раствор NaOH, KOH) или алкоголяты щелочных
металлов (C2H5ONa, С2Н5ОК и др.). Элиминированию
способствуют также повышение температуры
реакционной смеси и концентрации
реагентов. Благоприятное влияние
оказывает увеличение числа заместителей
у электрофильного атома углерода.
Поэтому особенно легко элиминирование
протекает в ряду третичных галогеналканов.
Если в молекуле галогеналкана имеется
несколько альтернативных путей отщепления
галогеноводорода, то преимущественно
реализуется из них тот, при котором
двойная связь образуется у наиболее
замещенного атома углерода; то есть
вместе с галогеном уходит водород от
наименее гидрогенизированного соседнего
атома углерода. Эта закономерность
получила название «правило
Зайцева»:
 Аналогично
нуклеофильному замещению элиминирование
галогеналканов может протекать по
бимолекулярному (E2) и мономолекулярному
(E1) механизмам. Механизм E2 имеет большое
сходство с механизмом SN2. Механизм
представлен следующей схемой:
Аналогично
нуклеофильному замещению элиминирование
галогеналканов может протекать по
бимолекулярному (E2) и мономолекулярному
(E1) механизмам. Механизм E2 имеет большое
сходство с механизмом SN2. Механизм
представлен следующей схемой:
 Как
видно, реакция по механизму E2 аналогично
механизму SN2 идет в одну стадию с
образованием переходного состояния, в
формировании которого принимают участие
молекулы двух реагентов. Поэтому скорость
такой реакции зависит от концентрации
обоих реагентов и описывается кинетическим
уравнением второго порядка. Процессы
разрыва и образования связей в переходном
состоянии происходят синхронно. Различие
между механизмами SN2 и E2 состоит в том,
что в механизме SN2 частица с неподеленной
парой электронов или несущая отрицательный
заряд атакует электрофильный атом
углерода молекулы галогеналкана,
действуя при этом как нуклеофил, а в
механизме E2 она атакует атом водорода
при β-углеродном атоме, действуя как
основание. Поэтому процессы SN2 и E2
являются конкурирующими. Наиболее легко
по механизму E2 происходит элиминирование
в ряду первичных алканов. Как механизм
E2 сходен с механизмом SN2, так и механизм
E1 имеет большое сходство с SN1 и конкурирует
с ним. Реакция, идущая по механизму E1,
представляет собой двухстадийный
процесс. На стадии I, как и в реакциях по
механизму SN1, под влиянием растворителя
происходит ионизация молекулы
галогеналкана с образованием карбокатиона.
Процесс ионизации идет медленно и
определяет скорость реакции в целом.
На стадии II образовавшийся карбокатион
стабилизируется, отщепляя протон от
β-углеродного атома с образованием
алкена:
Как
видно, реакция по механизму E2 аналогично
механизму SN2 идет в одну стадию с
образованием переходного состояния, в
формировании которого принимают участие
молекулы двух реагентов. Поэтому скорость
такой реакции зависит от концентрации
обоих реагентов и описывается кинетическим
уравнением второго порядка. Процессы
разрыва и образования связей в переходном
состоянии происходят синхронно. Различие
между механизмами SN2 и E2 состоит в том,
что в механизме SN2 частица с неподеленной
парой электронов или несущая отрицательный
заряд атакует электрофильный атом
углерода молекулы галогеналкана,
действуя при этом как нуклеофил, а в
механизме E2 она атакует атом водорода
при β-углеродном атоме, действуя как
основание. Поэтому процессы SN2 и E2
являются конкурирующими. Наиболее легко
по механизму E2 происходит элиминирование
в ряду первичных алканов. Как механизм
E2 сходен с механизмом SN2, так и механизм
E1 имеет большое сходство с SN1 и конкурирует
с ним. Реакция, идущая по механизму E1,
представляет собой двухстадийный
процесс. На стадии I, как и в реакциях по
механизму SN1, под влиянием растворителя
происходит ионизация молекулы
галогеналкана с образованием карбокатиона.
Процесс ионизации идет медленно и
определяет скорость реакции в целом.
На стадии II образовавшийся карбокатион
стабилизируется, отщепляя протон от
β-углеродного атома с образованием
алкена:
 Акцептором
протона часто служит сам растворитель,
например вода, поэтому протекающая по
механизму E1 реакция обычно не требует
присутствия основания как реагента. В
реакции элиминирования по механизму
E1 наиболее легко вступают третичные
галогеналканы.
Акцептором
протона часто служит сам растворитель,
например вода, поэтому протекающая по
механизму E1 реакция обычно не требует
присутствия основания как реагента. В
реакции элиминирования по механизму
E1 наиболее легко вступают третичные
галогеналканы.