

Полиядерные арены с изолированными циклами
Из углеводородов с изолированными бензольными ядрами наибольший интерес представляют ди- и трифенилметаны, а также бифенил.

Реакции электрофильного замещения
Экспериментальные данные показывают, что бифенил в реакциях электрофильного замещения более активен, чем бензол. Электрофильные реагенты атакуют орто- и пара-положения фенильных колец, причем преимущественно пара-положение (орто-атомы водорода одного кольца пространственно экранируют орто-положения другого кольца, что затрудняет их атаку электрофилом).
Строение -комплекса, образующегося после атаки молекулы бифенила электрофилом можно представить в виде следующего набора граничных структур:

Образование резонансных структур (IY), (Y) и (YI) должно быть затруднено по следующим причинам: 1) оба кольца в них должны быть компланарны, что приведет к довольно сильному взаимному отталкиванию орто-атомов водорода; 2) нарушается ароматическая система второго бензольного кольца, что энергетически не выгодно.С другой стороны, резонансная структура (II) предполагает определенное участие второго кольца в делокализации положительного заряда в -комплексе. Наиболее вероятно, что в этом случае проявляется положительный индуктивный, а не мезомерный (условие образования резонансных структур IY, Y и YI) эффект второго бензольного кольца.
Бифенил легко галогенируется, сульфируется, нитруется.

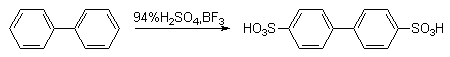

При переходе от бифенила к флуорену, в котором оба бензольных кольца строго компланарны и их взаимное влияние более ярко выражено, скорость реакций электрофильного замещения резко возрастает. При этом, как правило, образуются 2-замещенные флуорены.
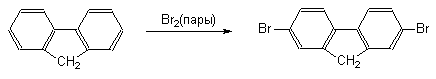

В ди- и трифенилметанах бензольные кольца полностью автономны и в реакциях электрофильного замещения они ведут себя подобно монозамещенным бензолам, содержащим объемные алкильные заместители.
Реакции метиленовой и метиновой групп в ди- и триарилметанах
Особенности химического поведения ди- и трифенилметанов проявляются в свойствах С-Н связи алифатической ("метановой") части молеулы. Легкость гетеро- или гомолитического разрыва этой связи зависит прежде всего от возможности делокализации возникающего положительного или отрицательного заряда (в случае гетеролитического разрыва) или неспаренного электрона (в случае гомолитического разрыва). В ди- и особенно в трифенилметановой системе возможность такой делокализации исключительно велика.
Рассмотрим способность фенилированных метанов к диссоциации С-Н связи с отщеплением протона ( СН-кислотность ). Сила СН-кислот, как и обычных протонных кислот, определяется устойчивостью, а следовательно и легкостью образования, соответствующих анионов (в рассматриваемом случае - карбанионов). Устойчивость и легкость образования анионов, в свою очередь, определяются возможностью делокализации в них отрицательного заряда. Каждое бензольное ядро, связанное с бензильным атомом углерода, может принимать участие в делокализации возникающего на нем отрицательного заряда, что можно представить с помощью резонансных структур:

Д ля
дифенилметана можно изобразить уже
семь граничных структур:
ля
дифенилметана можно изобразить уже
семь граничных структур:
а для трифенилметана – десять. Поскольку с числом возможных граничных структур растет способность к делокализации, ди- и особенно трифенилметил-анионы должны обладать особой устойчивостью. В связи с этим можно ожидать, что СН-кислотность метанов будет увеличиваться с увеличением числа фенильных колец, которые могут принимать участие в делокализации заряда на центральном атоме углерода, т.е. возрастать в ряду:
СН4 < С6Н5СН3 < (С6Н5)2СН2 < (С6Н5)3СН
Значения pKa указанных углеводородов, определенные специальными методами, подтверждают это предположение. Дифенилметан (pKa 33) по кислотности приблизительно равен аммиаку, а трифенилметан (pKa 31.5) - трет-бутанолу и более чем в 1010 раз превосходит по кислотности метан (pKa~ 40).
О![]() крашенный
в вишневый цвет трифенилметилнатрий
обычно получают восстановлением
трифенилхлорметана амальгамой натрия.
крашенный
в вишневый цвет трифенилметилнатрий
обычно получают восстановлением
трифенилхлорметана амальгамой натрия.
В![]() отличие от обычных СН-связей sp3-гибридного
атома углерода, бензильная С-Н связь
три-(пара-нитрофенил)метана
гетеролитически расщепляется уже
спиртовой щелочью.
отличие от обычных СН-связей sp3-гибридного
атома углерода, бензильная С-Н связь
три-(пара-нитрофенил)метана
гетеролитически расщепляется уже
спиртовой щелочью.
В последнем случае в делокализации отрицательного заряда в анионе помимо трех бензольных ядер дополнительно участвуют три нитрогруппы.
Другой вид гетеролитического расщепления бензильной СН-связи - отрыв гидрид-аниона с образованием соответствующих карбокатионов бензильного типа:
П оскольку
бензольные ядра способны стабилизировать
как положительный, так и отрицательный
заряды, фенилированные метаны по
гидридной подвижности водорода в
алифатической части составят тот же
ряд, что и по протонной подвижности:
оскольку
бензольные ядра способны стабилизировать
как положительный, так и отрицательный
заряды, фенилированные метаны по
гидридной подвижности водорода в
алифатической части составят тот же
ряд, что и по протонной подвижности:
СН4< С6Н5СН3< (С6Н5)2СН2< (С6Н5)3СН.
Однако экспериментально сравнить легкость отрыва гидрид-аниона, как правило, бывает трудно, поскольку для осуществления такого отрыва обычно используют весьма активные кислоты Льюиса. Сравнительные оценки легко могут быть сделаны путем сопоставления подвижности галогена (обычно хлора) в условиях SN1 реакций, поскольку в этом случае, как и при отщеплении гидрид-аниона, стадией, определяющей скорость превращения, является образование соответствующего карбокатиона.
Ar-CR2-Cl ArCR2+ + Cl-; (R = H , Ar)
Действительно, оказалось, что в указанных условиях наибольшей подвижностью хлор обладает в трифенилхлорметане, а наименьшей - в бензилхлориде.
(C6H5)3C-Cl > (C6H5)2CH-Cl > C6H5CH2-Cl
Реакционная способность хлора в трифенилхлорметане напоминает таковую в хлорангидридах карбоновых кислот, а в дифенилметане - в аллилхлориде. Ниже приведены данные об относительных скоростях сольволиза хлоридов R-Cl в муравьиной кислоте при 25о С:
R-Cl + HCOOH R-O-C(O)H + HCl
R |
CH2=CH-CH2 |
C6H5-CH2 |
(CH3)3C |
(C6H5)2CH |
(C6H5)3C |
Относительные скорости |
0.04 |
0.08 |
1 |
300 |
3.106 |
С равнительная
устойчивость трифенилметильного
(тритильного) катиона подтверждается
также многими другими экспериментальными
данными. Примером может служить легкость
образования его солей с ненуклеофильными
анионами, растворы которых в полярных
апротонных растворителях электропроводны
(а, значит, имеют ионное строение) и
характерно окрашены в желтый цвет:
равнительная
устойчивость трифенилметильного
(тритильного) катиона подтверждается
также многими другими экспериментальными
данными. Примером может служить легкость
образования его солей с ненуклеофильными
анионами, растворы которых в полярных
апротонных растворителях электропроводны
(а, значит, имеют ионное строение) и
характерно окрашены в желтый цвет:
О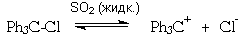 том же свидетельствует способность
трифенилхлорметана к диссоциации на
трифенилметил-катион и хлорид-анион в
растворе жидкого диоксида серы:
том же свидетельствует способность
трифенилхлорметана к диссоциации на
трифенилметил-катион и хлорид-анион в
растворе жидкого диоксида серы:
Устойчивость трифенилметильного катиона возрастает при введении в бензольные кольца электронодонорных групп (например, амино-, алкил- и диалкиламино-, гидроксильной, алкоксильной). Дальнейшее увеличение устойчивости карбокатиона приводит к ситуации, когда он становится устойчивым в водном растворе, то есть равновесие реакции
сдвинуто влево. ![]()
Подобные тритильные катионы окрашены. Примером может служить интенсивно окрашенный в фиолетовый цвет три(4-диметиламинофенил)метильный катион. Его хлорид применяют в качестве красителя под названием " кристаллический фиолетовый". В кристаллическом фиолетовом положительный заряд рассредоточен между тремя атомами азота и девятью атомами углерода бензольных ядер. Участие одного из трех пара-диметиламинофенильных заместителей в делокализации положительного заряда может быть отражено с помощью следующих граничных структур:
В се
трифенилметановые красители, содержащие
аминогруппы в бензольном кольце,
приобретают окраску в кислой среде,
которая способствуют возникновению
хиноидной структуры с протяженной
цепью сопряжения. Ниже приведены формулы
наиболее распространенных трифенилметановых
красителей.
се
трифенилметановые красители, содержащие
аминогруппы в бензольном кольце,
приобретают окраску в кислой среде,
которая способствуют возникновению
хиноидной структуры с протяженной
цепью сопряжения. Ниже приведены формулы
наиболее распространенных трифенилметановых
красителей.
(п-R2N-C6H4)2C+(C6H5)Cl- |
R = CH3 малахитовый зеленый |
R = C2H5 бриллиантовый зеленый |
|
R = H фиолетовый Дебнера |
|
(п-R2N-C6H4)3C+Cl- |
R = H парафуксин |
R= CH3 кристаллический фиолетовый |
А налогичное
влияние должны оказывать бензольные
ядра и на устойчивость трифенилметильного
радикала. Трифенилметильный радикал
может быть генерирован из соответствующего
хлорида действием цинка, меди или
серебра, которые в этом случае выступают
как доноры электрона.
налогичное
влияние должны оказывать бензольные
ядра и на устойчивость трифенилметильного
радикала. Трифенилметильный радикал
может быть генерирован из соответствующего
хлорида действием цинка, меди или
серебра, которые в этом случае выступают
как доноры электрона.
Т рифенилметильный
радикал достаточно устойчив и в
разбавленных растворах (в эфире, бензоле)
димеризуется лишь частично. При
димеризации возникает связь между
центральным углеродным атомом одного
радикала и пара-положением одного
из фенильных ядер другого радикала.
рифенилметильный
радикал достаточно устойчив и в
разбавленных растворах (в эфире, бензоле)
димеризуется лишь частично. При
димеризации возникает связь между
центральным углеродным атомом одного
радикала и пара-положением одного
из фенильных ядер другого радикала.
По-видимому один трифенилметильный радикал атакует наименее пространственно затрудненное место другого, Степень диссоциации таких димеров сильно зависит от природы арильных радикалов. Так, в 0.1 М растворе в бензоле при 25оС трифенилметильный радикал димеризован на 97%, а три-(4-нитрофенил)метильный не димеризуется вообще.