

Medical Genetics / ЛЕКЦИЯ ГБ и МФЗ 2021 ЛЕЧ 16х9
.pdfПрепараты для лечения наследственных болезней обмена
(зарегистрированы в России):
•2008 г. – Изифен (ФКУ)
•2008 г. – Альдуразим (МПС1)
•2008 г. – Элапраза (МПС2)
•2008 г. - Гемидон (Гомоцистинурия)
•2009 г – Глютаридон (Глютаровая ацидурия)
•2009 г – Наглазим (МПС6)
•2009 г - Реплагал (болезнь Фабри)
•2009 г – Орфадин (тирозинемия I типа)
•2009 г. - Миглустат (Ниманна-Пика, тип С) и другие

Профилактика НБО
•Ранняя постнатальная диагностика (идентификация) наследственных болезней обмена
•Диагностика гетерозиготного носительства
•Пренатальная (дородовая) диагностика и внутриутробная коррекция патологи плода
•Преклиническая (пресимптоматическая) диагностика
•Медико-генетическое консультирование

ЦЕЛЕСООБРАЗНОСТЬ ПРОВЕДЕНИЯ МАССОВОГО НЕОНАТАЛЬНОГО БИОХИМИЧЕСКОГО СКРИНИНГА НА НБО:
 Высокая частота встречаемости в популяциях Выявление больных детей на доклинической стадии Высокая чувствительность метода
Высокая частота встречаемости в популяциях Выявление больных детей на доклинической стадии Высокая чувствительность метода
Лечение начатое в первые дни жизни предотвращает развитие тяжелой клиники
Лечение дешевое, простое, очень эффективное
Высокая экономическая эффективность

ПЕРСПЕКТИВЫ: расширение неонатального скрининга (до 36 заболеваний)
•29 наследственный болезней обмена, выявляемых методом тандемной массспектрометрии (ТМС)
•2 наследственных заболевания обмена (галактоземия, дефицит биотинидазы), выявляемых флуориметрическим анализом
•3 заболевания (врожденный гипотиреоз, муковисцидоз, адрено-генитальный синдром) выявляемых иммуноферментным анализом
•2 наследственных заболевания (спинальная мышечная атрофия, первичные иммунодефициты), выявляемых молекулярно-генетическим методом.
24

Тандемная масс-спектрометрия
Аминоацидопатии |
Органические ацидурии |
|
Лейциноз (1:185000) |
Глютаровая ацидурия тип 1 (1:30000) |
|
Пропионовая ацидемия (1:50000) |
||
ФКУ (1:8000) |
||
Метилмалоновая ацидурия (1:48000) |
||
Тирозинемия тип1 (1:100000) |
||
Изовалериановая ацидурия (1:50000) |
||
Некетотическая гиперглицинемия (1:55000) |
||
|
||
Цитруллинемия (1:250000) |
|
Дефекты β-окисления
Недостаточность SCAD (недостаточность короткоцепочечной ацил-КоА дегидрогеназы жирных кислот)
Недостаточность MCAD (1:800) (недостаточность среднецепочечной ацил-КоА дегидрогеназы жирных кислот)
Недостаточность VLCAD (недостаточность длинноцепочечной ацил-КоА дегидрогеназы жирных кислот)
Недостаточность LCAHD (недостаточность митохондриального трифункционального белка ( недостаточность длиноцепочечной 3-гидроксиацил-КоА дегидрогеназы))
Недостаточность CPT1 (недостаточность карнитин-пальмитоилтрансферазы I) |
25 |
Недостаточность CPT2 (недостаточность карнитин-пальмитоилтрансферазы II) |
|
|
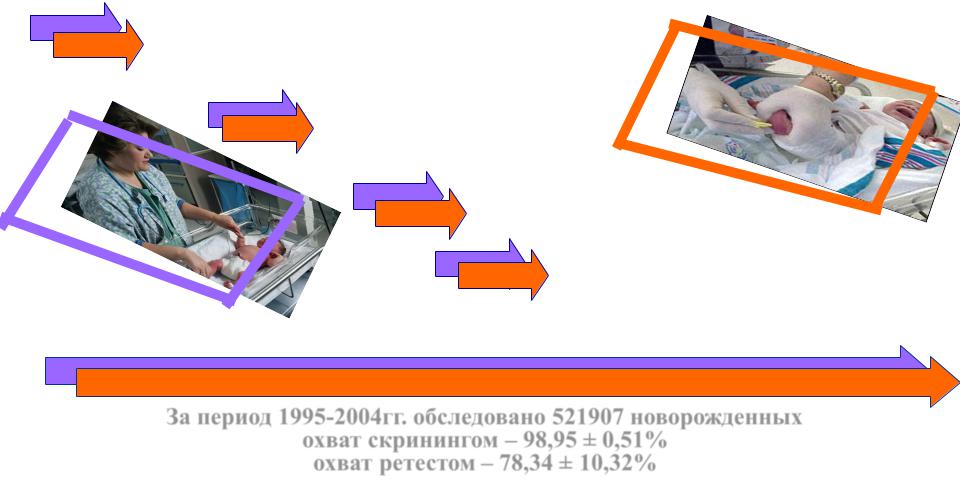
Алгоритм скрининга на НБО
У доношенных новорожденных кровь берется на 4 день жизни, при преждевременных родах – на 7 день
Доставка образцов в
лабораторию МГК не реже 1 раза в 3 дня
Лабораторные исследования 1-3
Вызов детей с высокими результатами до 10 дня жизни
диагноза до 12-14 дней жизни, наблюдение педиатра по месту
жительства
За период 1995-2004гг. обследовано 521907 новорожденных
охват скринингом – 98,95 ± 0,51% охват ретестом – 78,34 ± 10,32%
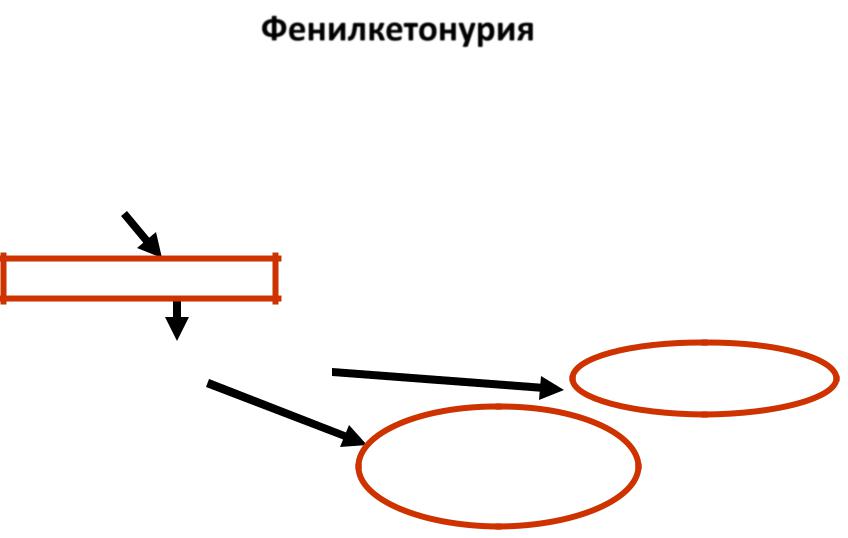
Фенилкетонурия
-наследственное заболевание, при котором отсутствует фенилаланин-4- гидроксилаза (один из ферментов, расщепляющих пищевые белки)
Пищевой белок
фенилаланин
Накопление в организме
фенилаланина
и его производных
Фенилаланин-4- гидроксилаза
 тирозин
тирозин
Метаболические
нарушения
Накопление в формирующейся нервной системе

ФЕНИЛКЕТОНУРИЯ
Особенностями ФКУ является:
1. Высокая частота болезни в европейских странах (1:4560 – Ирландия, 1:5600 – Белоруссия, 1:6700 – Германия, 1:8000 – Чехия,
1:6350 – Россия, 1:11800 – Москва).
2. Тяжелый исход болезни (необратимая умственная отсталость) в случае несвоевременной диагностики
(до первого полугодия жизни).
3. Высокая популяционная частота гетерозиготного носительства гена болезни – 1:50 человек.
В нашей стране проживают тысячи больных ФКУ, из
которых только около 2000 зарегистрированы официально

Частота ФКУ 1:6700
|
|
|
|
1: 7000 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
1:6200 |
|
|
|
|
|
|
||
|
|
|
|
1: 4500 |
|
|
||||
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
1: 5800 |
|
|
|
|
|
|
|
|
|
|
|
1: 9000 |
|
|
1:5500 |
|
|
|
||||
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
1: 10000
Краснодарский край - 1:8250 Ставропольский край - 1:13500 Ростовская область - 1:4000

|
Клиническая картина ФКУ |
|
Нарушение обмена |
|
Клиника |
|
|
|
Высокое содержание |
|
Поражение ЦНС (умственная отсталость, нарушения внимания) |
фенилаланина в крови и мозге |
|
|
|
|
|
Накопление фенилкетонов |
|
Необычный (затхлый, мышиный) запах мочи и пота |
|
|
Экземоподобные поражения кожи |
|
|
|
Дефицит тирозина |
|
Недостаток меланина (светлые кожа и волосы) |
|
|
Недостаток биоактивных аминов (серотонин, допамин) – |
|
|
судороги, тремор |
|
|
|
Манифестация происходит на первом году жизни,
вялость обычно в возрасте 2-6 мес. 
отсутствие интереса к окружающему
иногда повышенная раздражительность
беспокойство
срыгивание
мышечная гипо- и гипертония
судороги
признаки аллергического дерматита
характерный «мышиный» запах
гипопигментация кожи, волос, радужной оболочки глаз
отчетливо формируется задержка психомоторного развития; при отсутствии лечения развивается тяжелая умственная отсталость.