

Лекции / Kurs_lektsiy_po_patofiziologii_Ch_4_2018
.pdfСиндром Ди Джорджи (гипоили аплазия тимуса)
Причиной является делеция 22 хромосомы, которая приводит к нарушению формирования 3-4-го жаберных карманов во внутриутробном периоде. Характеризуется нарушением развития тимуса, паращитовидных желез, дуги аорты и структур лица. Гипоплазия тимуса сочетается с недоразвитием или полным отсутствием паращитовидных желез, что сопровождается развитием гипокальциемии и судорогами. Кроме этого типичными являются пороки развития лица, ушей, сердца и аорты.
ИД является следствием нарушения или полного отсутствия вилочковой железы. При этом количество T-лимфоцитов в крови, в тимус зависимых зонах лимфатических узлов и селезенки снижено. Нарушен клеточный иммунный ответ, поэтому больные подвержены вирусным, грибковым и протозойным инфекциям, а также заболеваниям, вызванным внутриклеточными бактериями (возбудителями туберкулеза, лепры, бруцеллеза и др.). Гуморальные иммунные реакции сохранны. Лечение симптоматическое.
Алимфоцитоз (синдром Незелофа) наследуется аутосомно-
рецессивно. Тимус недоразвитый и не способен поддерживать Т- клеточную дифференцировку, поэтому развивается количественная и качественная недостаточность Т-лимфоцитов. Проявляется задержкой роста, развитием сепсиса с появлением гнойных очагов в коже, легких и других органах. Характерны опустошение тимусзависимых зон в лимфоузлах, развитие лимфопении за счет уменьшения Т- клеток, снижение реакции бласттрансформации лимфоцитов. Содержание иммуноглобулинов в периферической крови нормальное. Лечение симптоматическое.
Иммунный дефицит с тромбоцитопенией и экземой (синдром Вискотта-Олдрича)
Наследуется рецессивно сцеплено с Х-хромосомой. Основу со-
ставляет дефект генов WAS (Wisskott-Aldrich syndrome) и WASP.
Синдром характеризуется нарушением активации CD4+ и CD8+- клеток, уменьшением продукции Ig М к капсулярным бактериям (не вырабатываются антитела к полисахаридам). Концентрация Ig G в сыворотке нормальная на фоне повышения содержания Ig А и Ig Е. В периферической крови, как правило, нормальный уровень Т- и В- лимфоцитов, но может быть лимфоцитопения и низкая активность Т- лимфоцитов в ответ на полисахаридные АГ.
19
Диагностируется у мальчиков в раннем возрасте (с 6-ти месячного возраста). Заболевание начинается с геморрагических проявлений из-за тромбоцитопении: диарея с кровью, петехии на коже и слизистых. С первого года жизни появляется прогрессирующая экзема, склонная к переходу в нейродермит.
Противоинфекционная устойчивость постепенно снижается, часто развиваются отиты, ангины, назофарингиты, конъюктивиты. В итоге заболевание, как правило, заканчивается развитием фатальной инфекции или лимфопролиферативным процессом (с образованием лимфом). Лечение симптоматическое.
Луи-Бар синдром (атаксия – телеангиэктазия)
Это – комплексное заболевание иммунной, нервной и эндокринной систем, наследуемое аутосомно-рецессивно. Основу составляет дефект гена АТМ, кодирующего ДНК-топоизомеразу.
Атаксия связана с прогрессирующей дегенерацией клеток Пуркинье в мозжечке и обнаруживается уже в 2-4-месячном возрасте. Телеангиоэктазии конъюнктивы, кожи, слизистых появляются к 3-6 годам. Иммунные нарушения состоят в развитии гипоплазии тимуса, лимфатических узлов, селезёнки, миндалин, функциональной недостаточности T-лимфоцитов (число T-лимфоцитов у большинства пациентов нормальное). Уровни Ig A, Ig E, Ig G2 снижены. Типично развитие эндокринологических расстройств: гипоплазии или аплазии яичников, снижение толерантности к глюкозе, низкорослость.
Иммунодефицит проявляется инфекционным синдромом и высокой частотой новообразований (лимфомы и карциномы). Лечение симптоматическое.
4.1.1.3.Первичные дефициты системы фагоцитов
Хроническая гранулематозная болезнь характеризуется наличи-
ем генетически детерминированных метаболических дефектов в фагоцитах (макрофагах, нейтрофилах), которые поглощают патогены, но не могут их расщепить и уничтожить. Фагоцит становится резервуаром для сохранивших жизнеспособность микроорганизмов, которые могут выходить из фагоцитов и стимулировать развитие гранулем. Данная патология имеет несколько генетических дефектов, которые связаны с НАДФH-оксидазной системой генерации активных форм кислородных радикалов. Лечение заключается в антибиотико-
20
терапии. В редких случаях прибегают к трансплантации костного мозга.
Синдром Чедиака-Хигаси наследуется аутосомно-рецессивно, но точный генетический дефект неизвестен. Синдром характеризуется нарушением структуры внутриклеточных гранул фагоцитов и меланоцитов. Гранулы сливаются, образуя крупные, но функционально недееспособные гранулы. Проявлениями являются рекуррентные бактериальные инфекции, частичный глазокожный альбинизм (слившиеся гранулы меланоцитов не содержат меланина), склонность к кровоточивости, патологические проявления со стороны нервной системы (в нейронах также могут сливаться везикулы). Лечение симптоматическое.
Болезни с дефицитом молекул адгезии лейкоцитов (LAD – leucocyte adhesion deficiency) – очень редкая прижизненная патология, имеющая два клинически неразличимых варианта LAD-1 и LAD-2. При LAD-1 нарушен синтез интегринов, обеспечивающих адгезию лейкоцитов к эндотелиоцитам, агрегацию нейтрофилов, хемотаксис лейкоцитов, фагоцитоз, адгезию Т-лимфоцитов к АПК, В-лимфоцитам и клет- кам-мишеням. При LAD-2 генетический дефект другой: на лейкоцитах отсутствует лиганд для взаимодействия с Е- и Р-селектинами на активированном эндотелии, что приводит к нарушению миграции лейкоцитов в очаг инфекции. Проявляются в плохом заживлении ран и развитии рекуррентных бактериальных, грибковых, вирусных и паразитарных инфекциях. Лечение симптоматическое.
4.1.1.4. Дефициты системы комплемента
Описаны практически для всех компонентов комплемента человека (C1q, C1r, C1s, C2-C9, пропердина) и подразделяются на:
1)дефекты ингибиторов системы комплемента;
2)генетически детерминированные дефекты строения фракций системы комплемента;
3)наследственный абсолютный дефицит определенных фракций системы комплемента.
Дефект синтеза ингибитора С1-эстеразы (наследственный ан-
гионевротический отек) проявляется отеком в области гортани или желудочно-кишечного тракта, в основе которых лежат пароксизмы неограниченной активации системы комплемента.
21
Дефицит компонентов С1-С4 может быть обусловлен недостаточным синтезом или избыточным их катаболизмом и проявляется в болезнях иммунных комплексов – системных васкулитах и повреждениях почек (системная красна волчанка). При дефиците фракции С3 также отмечается недостаточность иммунной адгезии фагоцитов, т.к. С3b является опсонином, а также и повышенная восприимчивость к инфекции, вызываемой Neisseria spp.
Семейная дисфункция фракции комплемента С5 встречается при синдроме Лейнера, при котором больные страдают от системного себорейного дерматита, тяжелого поноса и частых бактериальных инфекций при инвазии во внутреннюю среду преимущественно грамотрицательных микроорганизмов.
4.1.1.5. Принципы профилактики и терапии первичных ИД
Принципы профилактики первичных иммуннодефицитов
Среди населения, особенно среди беременных, целесообразно проводить профессиональное медико-генетическое консультирование. При выявлении внутриутробной и постнатальной недостаточности иммунной системы, отмечаемой при разнообразных токсикозах беременных, необходимо предпринять следующие действия: 1) провести роды при помощи кесарева сечения; 2) поместить новорождённого в стерильные условия; 3) обеспечить гнотобиотическое содержание ребёнка; 4) резко ограничить широкое использование антибиотиков, цитостатиков и других лекарственных средств; 5)исключить контакт с бытовыми и промышленными ядами, т.к. они способны вызвать развитие тератогенных нарушений и мутаций.
Принципы терапии первичных иммуннодефицитов
Прежде чем начать конкретное лечение ИД, необходимо выяснить его тип и степень, а также уровень повреждения иммунной системы и механизмы развития иммунной недостаточности. Для лечения ИД используют преимущественно этиотропное и патогенетическое лечение.
Этиотропная терапия направлена на устранение или ослабление действия патогенных факторов, в том числе лекарственных, бытовых и производственных, способных вызвать повреждения различных
22
звеньев иммунной системы, а также на повышение устойчивости (резистентности) организма к действию этих факторов.
Патогенетическая терапия. При комбинированном ИД восстановительное патогенетическое лечение сводится к проведению трансплантации эмбриональных тканей костного мозга, вилочковой железы, печени, в том числе стволовых клеток, части совместимого костного мозга либо отдельных его фракций.
При преимущественно гуморальном ИД, а также при некоторых комбинированных формах ИД целесообразно проводить заместительную патогенетическую терапию путём внутривенного введения совместимой свежей плазмы крови, содержащей либо многие иммуноглобулины, либо конкретные недостающие иммуноглобулины, в том числе, γ-глобулины. Также назначают антитоксические, антигриппозные и другие сыворотки.
Возможна активная иммунизация соответствующими вакцинами. Целесообразно проводить фармакологическую коррекцию эффекторного звена иммунной системы иммуномодуляторами, медиаторами иммунной системы, гормонами, адаптогенами, а также соответствующую противомикробную и противогрибковую терапию.
Необходимо выводить из организма разные ингибирующие факторы, связывающие АТ и блокирующие эффекты иммунокоррекции, путём проведения гемосорбции, гемодиализа, плазмо- и лимфофереза.
При избирательной недостаточности Ig A не рекомендуют введение плазмы и комплекса иммуноглобулинов.
При избирательном (или преимущественном) клеточном иммунодефиците противопоказано переливание свежей крови и препаратов крови.
При одновременном наличии тромбоцитопении и ИД необходимо избегать проведения хирургических и стоматологических вмешательств, а также внутривенных и внутримышечных инъекций.
23
4.1.2.Вторичные иммунодефициты
Вторичные ИД – нарушения иммунной системы, которые развиваются в позднем постнатальном периоде или у взрослых и которые не являются генетически детерминированными; клинически проявляются рецидивирующими инфекциями, плохо поддающимися традиционным методам лечения (табл.1).
Таблица 1 Причины и механизмы развития вторичных иммунодефицитов
|
Причины ИД |
Механизм формированиия ИД |
||
Белково-калорийное голода- |
Гипогаммаглобулинемия |
|
||
ние |
|
|
|
|
Дефицит железа |
|
Нарушение функции Т-лимфоцитов |
||
Постинфекционный |
(вирус |
Гиперстимуляция иммунной системы |
||
Эпштейна-Барр, цитомега- |
суперАГами патогенов |
и массовая |
||
ловирусы, герпесвирусы че- |
поликлональная гибель (апоптозом) |
|||
ловека 6-го и 7-го типов (и |
активированных лимфоцитов |
|||
др.) |
|
|
|
|
Болезнь Ходжкина |
|
Нарушение функции Т-лимфоцитов |
||
|
|
|||
Множественная миелома |
Нарушение синтеза иммуноглобули- |
|||
|
|
|
нов |
|
Лимфома или лимфолейкоз |
Снижение количества |
нормальных |
||
|
|
|
лимфоцитов |
|
Поздние |
стадии |
злокаче- |
Снижение функции Т-лимфоцитов, |
|
ственных опухолей |
|
др. неизвестные механизмы |
||
Опухоли тимуса |
|
Гипогаммаглобулинемия |
|
|
Хроническая почечная недо- |
Неизвестен |
|
||
статочность |
|
|
|
|
Сахарный диабет |
|
Неизвестен |
|
|
Вызванный лекарствами им- |
Угнетение лимфопоэза |
|
||
мунодефицит (кортикосте- |
|
|
||
роиды, |
противоопухолевые |
|
|
|
препараты) |
|
|
|
|
ВИЧ-инфекция (СПИД) |
Снижение количества Т-лимфоцитов, |
|||
|
|
|
особенно Т-хелперов |
|
|
|
|
24 |
|
4.1.2.1.Синдром приобретенного иммунодефицита (СПИД)
Он вызывается вирусом-возбудителем, который был выделен в 1983 г. Является ретровирусом, имеющим фермент «обратная транскриптаза», которая при проникновении вируса в клетку-мишень обеспечивает синтез ДНК по матрице вирусной РНК. Другой вирусный фермент, интеграза, встраивает вирусную ДНК (провирус) в геном человека. С участием вирусной ДНК происходит синтез мРНК (трансляция белков вируса и синтез геномной РНК вируса).
Оболочка вируса – мембрана клетки человека, в которую встроены оболочечные белки вируса gpl20 (самый наружный) и gp41 (трансмембранный).
Пути заражения ВИЧ: парентеральное введение крови или продуктов крови, слизистые контакты, трансплацентарно, грудное вскармливание.
Основными клетками-мишенями вируса являются клетки, несущие на мембране CD4 молекулу и рецепторы для хемокинов семейства «СС» (так называемый рецептор CC-CKR5). Вирус инфицирует нейроны, CD4+ Т-лимфоциты, клетки эндотелия, дендритные клетки, моноциты, гистиофаги, фибробласты, В-лимфоциты, CD8+ Т- лимфоциты, промиелоциты, мегакариоциты, хондроциты, стволовые кроветворные клетки.
Воснове СПИДа лежит выраженная иммуносупрессия, первично вызванная нарушением клеточно-опосредованных иммунных реакций. Выделяют несколько механизмов иммунодепрессии при ВИЧинфекции (рис. 4).
ВИЧ оказывает прямое цитопатогенное действие на CD4+ Т- лимфоциты, вызывая их гибель. Кроме этого белок gpl20 конкурентно блокирует корецептор CD4, а также, связываясь с неинфицированными Т-хелперами, превращает их в мишень для цитотоксических лимфоцитов и антителозависимой клеточной цитотоксичности. Суперантигены ВИЧ индуцируют поликлональную активацию и апоптоз Т-лимфоцитов. ВИЧ стимулирует образование синцития.
Вмакрофагах ВИЧ угнетает хемотаксис, ухудшает презентацию АГ, ингибирует синтез молекул MHC I, нарушает фагоцитоз, опосредованный через Fc-peцептор, снижает эффективность всех бактерицидных механизмов.
25
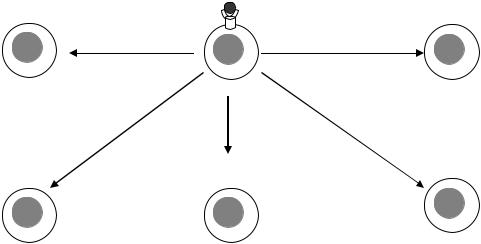
На уровне CD8+ Т-лимфоцитов ВИЧ первоначально вызывает лимфоцитоз с постепенным ухудшением функционирования цитотоксических лимфоцитов. Также значительно нарушается функция
NК.
Снижение |
ответа на |
|
|
растворимые АГ |
|
||
Снижение |
|
секреции |
|
регуляторных |
лимфо- |
ВИЧ |
|
|
|
|
|
кинов
CD4 |
CD4 |
CD8 |
NK |
Снижение специ- |
Снижение киллинга |
фической цитоток- |
опухолевых клеток |
сичности |
|
Снижение цитотоксичности, хемотаксиса, секреции IL-1, нарушение процессинга и презентации АГа
макрофаг
B
Нарушение продукции Ig в ответ на новые АГ
Рис. 4. Эффекты действия ВИЧ
ВИЧ вызывает поликлональную активацию В-лимфоцитов с развитием гипериммуноглобулинемии (Ig Gl, Ig G3, Ig A, Ig E): количество антител растет, но при этом способность к индукции антигенспецифичного гуморального ответа снижается. Кроме того, описан феномен усиления инфекции антителами, когда противовирусные антитела, связывая вирусы в комплексы, помогают им инфицировать клетки, имеющие Fc-рецепторы.
ВИЧ также поражает стволовые кроветворные клетки и тимоциты, ингибируя кроветворение и усугубляя иммунодефицит.
Клинически в развитии ВИЧ-инфекции можно выделить несколько периодов (по манифестации индикаторных болезней и числу CD4+ Т-лимфоцитов в периферической крови).
Острая фаза (стадия сероконверсии, виремии) длится несколько недель или месяцев после инфицирования. Клинически проявляется в
26
70 % случаев неспецифическим гриппоподобным синдромом. В крови обнаруживают вирус и вирусные АГ. Специфические AT появляются в крови через 3-6 мес после заражения. Тогда же в организме появляются вирус-специфические CD8+ цитотоксические Т- лимфоциты.
Гриппоподобный синдром саморазрешается и наступает многолетний бессимптомный период (до 10-12 лет). Больной остаётся сероположительным при отсутствии симптомов либо при их минимальной выраженности. Однако репликация вируса продолжается, прогрессивно уменьшается количество CD4+ клеток при сохраненной активности CD8+ цитотоксических лимфоцитов.
Симптоматическая фаза (2-3 года) характеризуется развитием оппортунистических инфекций, неоплазм на фоне прогрессирующего уменьшения CD4+ клеток. Возможно развитие саркомы Капоши, кандидозов и лейкоплакий слизистых оболочек, пневмоцистной пневмонии, токсоплазмоза с частым поражением головного мозга, заболеваний, вызванных микобактериями, цитомегаловирусами, криптококками (пневмонии, менингиты), герпес-вирусами.
Сокращение числа СD4+-лимфоцитов до 50 / мм3 и ниже приводит к полной дисфункции иммунной системы.
4.1.2.2. Принципы лечения СПИДа
Принципы лечения ВИЧ-инфекции основаны на политерапии, т.е. на применении комплекса последовательно назначаемых, различных по строению, механизмам и эффектам действия следующих средств:
−нейтрализующих вирус СПИДа;
−предотвращающих проникновение вируса в клетки организма;
−ингибирующих ранние этапы интеграции вирусной ДНК в геном клетки организма-хозяина;
−действующих в постинтегральной фазе жизни вирусных частиц;
−влияющих на отпочковывание (образование) вирусных частиц;
−действующих на заражённые вирусом клетки организма;
−активирующих или модулирующих различные звенья иммунной системы;
−обладающих способностью подавлять возбудители вторичной инфекции (например, другие вирусы и различные патогенные бактерии, грибы, простейшие и паразиты);
27
−ослабляющих расстройства различных физиологических и функциональных систем;
−активизирующих механизмы защиты, компенсации и приспособления.
4.2.Ииммунологическая толерантность организма
Иммунологическая толерантность организма – отсутствие иммунного ответа организма на определенный (ые) антиген (ы).
АГ, вызывающие иммунологическую толерантность, называются толерогены. Иммунологическая толерантность специфична, т.е. развивается к строго определенным АГ. Иммунологическая толерантность может быть поливалентной (ко всем антигенным детерминантам данного АГ) и моновалентной или расщепленной (избирательная невосприимчивость отдельных антигенных детерминант).
Выделяют физиологическую, патологическую и индуцированную иммунологическую толерантность.
4.2.1.Физиологическая (естественная) толерантность
Физиологическая (естественная) толерантность – терпи-
мость иммунной системы к АГ собственного организма.
Выделяют несколько механизмов естественной толерантности:
1)клонально-селекционный,
2)«изоляционный»,
3)анергия Т-лимфоцитов, не подвергшихся костимуляции («клональная анергия»),
4)апоптоз лимфоцитов, активизированных эндогенными АГ («клональная делеция»),
5)ликвидация аутоагрессивных Т-лимфоцитов в тимусе («центральная селекция»),
6)депрессия Т-киллеров Т-супрессорами.
4.2.1.1.Клонально-селекционный механизм
Клонально-селекционная теория (Ф. Бернет и Ф. Фенне) утверждает, что в организме существует гетерогенная популяция лимфоцитов, которые могут обеспечить ответ на любой АГ. Численность данной популяции составляет приблизительно 104 лимфоцитов. Каж-
28