

6. Понятие и классификация врожденных пороков развития.
Врожденными пороками развития называют такие структурные нарушения, которые возникают до рождения (в пренатальном онтогенезе), выявляются сразу или через некоторое время после рождения и вызывают нарушение функции органа. Последнее отличает врожденные пороки развития органов от аномалий, при которых нарушение функции обычно не наблюдается. Врожденные пороки развития являются причиной приблизительно 20% смертей в неонатальном периоде, а также занимают значительное место в практике акушерства и гинекологии, медицинской генетике детской хирургии и ортопедии, патологической анатомии. В связи с этим знания по вопросам профилактики, этиологии, патогенеза, лечения и прогнозирования врожденных пороков развития имеют большое значение.
Знакомство с закономерностями и механизмами нормального морфогенеза в процессе эмбрионального развития позволяет понять, какого рода нарушения могут привести к возникновению пороков. Кроме того, пороки развития представляют собой как бы природные эксперименты, обнаруживающие скрытые от глаз процессы. Таким образом, сами эти пороки представляют собой материал для научных поисков и обобщений. Примером могут служить такие врожденные пороки развития, которые напоминают некоторые черты строения, свойственные другим видам взрослых позвоночных животных или их зародышам. Они позволяют осознать теснейшую эволюционно-биологическую связь человека с животными и использовать ее для иллюстрации закономерностей макроэволюции, а также для формирования естественноисторического взгляда на возникновение и развитие человека.
Существует несколько различных критериев, на основе которых классифицируют врожденные пороки развития. Основные из них следующие: причина, стадия, на которой проявляется воздействие, последовательность их возникновения в организме, распространенность и локализация. Мы дополнительно обращаем внимание на филогенетическую значимость и нарушения основных клеточных механизмов, приводящие к пороку развития.
В зависимости от причины все врожденные пороки развития делят на наследственные, экзогенные (средовые) и мультифакториальные.
Наследственными называют пороки, вызванные изменением генов или хромосом в гаметах родителей, в результате чего зигота с самого возникновения несет генную, хромосомную или геномную мутацию. Генетические факторы начинают проявляться в процессе онтогенеза последовательно, путем нарушения биохимических, субклеточных, клеточных, тканевых, органных и организменных процессов. Время проявления нарушений в онтогенезе может зависеть от времени вступления в активное состояние соответствующего мутированного гена, группы генов или хромосом. Последствия генетических нарушений зависят также от масштаба и времени проявления нарушений.
Экзогенными называют пороки, возникшие под влиянием тератогенных факторов (лекарственные препараты, пищевые добавки, вирусы, промышленные яды, алкоголь, табачный дым и др.), т.е. факторов внешней среды, которые, действуя во время эмбриогенеза, нарушают развитие тканей и органов.
Историческими вехами являются работы Ц. Стоккарта в начале XX в., впервые показавшего тератогенное действие алкоголя, и работы офтальмолога Н. Грегга, открывшего тератогенное действие вируса краснухи (1941). Очень страшное событие имело место в 1959—1961 гг., когда после применения беременными талидомида в ряде стран Запада родились несколько десятков тысяч детей с тяжелыми врожденными пороками.
Поскольку средовые экзогенные факторы в конечном итоге оказывают влияние на биохимические, субклеточные и клеточные процессы, механизмы возникновения врожденных пороков развития при их действии такие же, как при генетических причинах. В результате фенотипическое проявление экзогенных и генетических пороков бывает весьма сходным, что обозначается термином фенокопия. Для выявления истинных причин возникновения пороков в каждом конкретном случае следует привлекать множество различных подходов и критериев.
Мультифакториальными называют пороки, которые развиваются под влиянием как экзогенных, так и генетических факторов. Вероятно, скорее всего бывает так, что экзогенные факторы нарушают наследственный аппарат в клетках развивающегося организма, а это приводит по цепочке ген — фермент — признак к фенокопиям. Кроме того, к этой группе относят все пороки развития, в отношении которых четко не выявлены генетические или средовые причины.
Установление причины врожденных пороков имеет большое прогностическое значение для носителя этих пороков и профилактическое — в отношении последующего потомства. В настоящее время медицинские генетики и патологоанатомы существенно продвинулись в области так называемого синдромологического анализа. Синдромологический анализ — это обобщенный анализ фенотипа больных с целью выявления устойчивых сочетаний признаков. Овладение им помогает в установлении причины возникновения пороков и основных патогенетических механизмов.
В зависимости от стадии, на которой проявляются генетические или экзогенные воздействия, все нарушения, происходящие в пренатальном онтогенезе, подразделяют на гаметопатии, бластопатии, эмбриопатии и фетопатии. Если нарушения развития на стадии зиготы
(гаметопатия) или бластулы (бластопатия) очень грубые, то дальнейшее развитие, видимо, не идет и зародыш погибает. Эмбриопатии (нарушения, возникшие в период от 15 сут до 8 нед эмбрионального развития) как раз составляют основу врожденных пороков, о чем уже говорилось выше. Фетопатии (нарушения, возникшие после 10 нед эмбрионального развития) представляют собой такие патологические состояния, для которых, как правило, характерны не грубые морфологические нарушения, а отклонения общего типа: в виде снижения массы, задержки интеллектуального развития, различных функциональных нарушений. Очевидно, что наибольшее клиническое значение имеют эмбриопатии и фетопатии.
В зависимости от последовательности возникновения различают первичные и вторичные врожденные пороки. Первичные пороки обусловлены непосредственным действием тератогенного фактора, вторичные — являются осложнением первичных и всегда патогенетически с ними связаны. Выделение первичных пороков из комплекса нарушений, обнаруженных у пациента, имеет большое значение для медико-генетического прогноза, поскольку риск определяется по основному пороку.
По распространенности в организме первичные пороки подразделяют на изолированные, или одиночные, системные, т.е. в пределах одной системы, и множественные, т.е. в органах двух систем и более. Комплекс пороков, вызванный одной ошибкой морфогенеза, называют аномаладом.
В основу классификации врожденных пороков, принятой Всемирной организацией здравоохранения (ВОЗ), положен анатомо-физиологический принцип (по месту локализации).
По клеточным механизмам, которые преимущественно нарушены при том или ином врожденном пороке развития, можно выделить пороки, возникшие в результате нарушения размножения клеток, миграции клеток или органов, сортировки клеток, дифференцировки, а также гибели клеток. Нарушение перечисленных клеточных механизмов может привести к слишком малым или, наоборот, слишком большим размерам органов или их частей, к недостаточному или, напротив, очень сильному рассасыванию тканей в органах, к изменению положения отдельных клеток, тканей или органов относительно других органов и тканей, к нарушениям дифференцировки, так называемым дисплазиям.
По филогенетической значимости можно все врожденные пороки развития разделить на филогенетически обусловленные и не связанные с предшествующим филогенезом, т.е. нефилогенетические.
Филогенетически обусловленными называют такие пороки, которые по виду напоминают органы животных из типа Хордовые и подтипа Позвоночные. Если они напоминают органы предковых групп или их зародышей, то такие пороки называют анцестральными (предковыми) или атавистическими. Примерами могут служить несращение дужек позвонков, шейные и поясничные ребра, несращение твердого нёба, персистирование висцеральных дуг и др. Если пороки напоминают органы родственных современных или древних, но боковых ветвей животных, то их называют аллогенными. Филогенетически обусловленные пороки показывают генетическую связь человека с другими позвоночными, а также помогают понять механизмы возникновения пороков в ходе эмбрионального развития.
+
Нефилогенетическими являются такие врожденные пороки, которые не имеют аналогов у нормальных предковых или современных позвоночных животных. К таким порокам можно
отнести, например, двойниковые уродства и эмбриональные опухоли, которые появляются в результате нарушения эмбриогенеза, не отражая филогенетических закономерностей.
7.Наследственные болезни с нетрадиционным наследованием: митохондриальные болезни, болезни импринтинга (синдром Прадера-Вилли, Энгельмана); экспансии тринуклеотидных повторов (синдром ломкой Х-хромосомы Мартина-Белла). Генотипическая и фенотипическая характеристика.
Как отмечалось выше, в последние годы обнаружен ряд новых закономерностей наследования признаков (нормальных и патологических), не соответствующих менделевским. К ним относятся: геномный импринтинг, экспансия триплетных повторов, митохондриальные болезни.
Совсем недавно у человека обнаружены однородительские дисомии. В норме ребенок наследует по каждой паре хромосом: одну хромосому от отца, другую - от матери. У индивидов с этим типом
наследования число хромосом по всем парам нормальное, однако одна пара представлена хромосомами от одного и того же родителя. Наиболее частый механизм возникновения однородительской дисомии у человека - редукция трисомии: в процессе гаметогенеза за счет нерасхождения хромосом возникает дисомия в гамете по определенной хромосоме, что при оплодотворении приводит к трисомии. По неясным пока причинам третья хромосома может элиминироваться на ранних стадиях дробления, а у зародыша останутся две хромосомы одного родителя. Следует отметить, что в целом частота возникновения однородительской дисомии материнского происхождения в 3 раза чаще отцовской. Если бы две копии генов, получаемых потомками от матери или от отца, функционировали в клетках одинаково, то никаких серьезных последствий в организме эти нарушения не вызывали бы. Однако оказалось, что некоторые хромосомы несут отдельные гены, экспрессия которых определяется полом передавшего их родителя. Это явление получило название феномена геномного импринтинга (от imprint - отпечаток).
Под геномным импринтингом понимают эпигенетический процесс, который дифференциально маркирует материнские и отцовские гомологичные хромосомы. В участках генома, подверженных импринтингу, экспрессируется (проявляется) только один из двух аллелей - отцовский или материнский, т.е. наблюдается моноаллельная экспрессия генов. Второй аллель, вследствие наличия на нем некоего отпечатка, импринтирован (выключен или подавлен) и не экспрессируется.
Такой способ регуляции генов свидетельствует о неэквивалентном вкладе родителей в геном потомков и приводит к разному фенотипическому проявлению мутаций у потомства, унаследованных от матери или отца.
У человека известно уже около 40 таких генов, и предполагается, что их число может достигать 200-500. Основную роль в процессе геномного импринтинга играет метилирование цитозиновых оснований ДНК с образованием 5-метилцитозина, способствующее выключению экспрессии генов с модифицированными нуклеотидами.
Так, известно, что в проксимальном участке хромосомы 15 имеются близко сцепленные, но противоположно импринтированные локусы, отвечающие за возникновение двух фенотипически разных синдромов - Прадера-Вилли и Энгельмана. Для синдрома
Прадера-Вилли, фенотипически проявляющегося умственной отсталостью, мышечной гипотонией, выраженным ожирением, гипогонадизмом, низким ростом и акромикрией, установлен кандидатный ген, ответственный за синтез полипептида N малого ядерного рибонуклеопротеина (SNRPN). Этот ген активно экспрессируется исключительно на отцовской хромосоме 15. Кандидатным геномом синдрома Энгельмана (синоним - синдром «счастливой куклы»), характеризующегося неадекватной счастливой улыбкой и глубокой умственной отсталостью с резкими кукольными судорожными движениями, является убиквитин - белковый лигазный ген (ИВЕЗА) E6-AP, который экспрессируется главным образом на материнской хромосоме 15.
Синдром Прадера-Вилли развивается при делеции участка 15 хромосомы отца (нет участка, который экспрессируется на отцовской хромосоме) или однородительской дисомии 15 хромосомы материнского происхождения (в этом случае гены импринтированы - выключены или подавлены).
Синдром Энгельмана («счастливой куклы») развивается при делеции участка 15 хромосомы матери или при однородительской дисомии 15 хромосомы отцовского происхождения.
В случае однородительской дисомии отсутствует потеря хромосомного материала, но нарушается нормальное метилирование ДНК, а следовательно, и транскрипция генов критического района, поэтому такое функциональное состояние равнозначно делеции.
В начале 1990-х гг. у человека был обнаружен новый тип мутаций, который до сих пор не зарегистрирован ни у одного вида млекопитающих. Он получил название динамических мутаций или мутаций экспансии (см. выше).
Общие характеристики этого класса болезней следующие:
Болезни с экспансией тринуклеотидных повторов представляют собой нейродегенеративные заболевания с поздним проявлением.
Отмечается прямая корреляция между числом тринуклеотидных повторов и тяжестью клинической картины.
Для болезней экспансии характерна генетическая антиципация - возрастание тяжести заболевания в последующих поколениях, что связано с тенденцией к возрастанию числа повторов у потомков.
Первое заболевание, при исследовании которого в 1991 г. был открыт феномен экспансии - синдром фрагильной (ломкой)
Х-хромосомы, или синдром Мартин-Белл. Проявления синдрома Мартин-Белл: умственная отсталость, аутизм, макроорхидизм (у взрослых), удлиненное лицо, прогнатия - выступающий подбородок, оттопыренные уши, пронзительная смешная речь, аномалии соединительной ткани, нарушение поведения.
Сейчас открыта целая группа болезней экспансии тринуклеотидных повторов.
Утверждение, что весь генетический материал человека находится в составе хромосом, не совсем верно, поскольку есть одно исключение - митохондриальный геном. Митохондриальная ДНК (мтДНК) представляет собой небольшую кольцевидную молекулу длиной 16 569 пар оснований. В отличие от ДНК ядерного генома она не связана с белками, имеет очень высокую «плотность генов» ввиду отсутствия интронов, содержит 13 генов, кодирующих белки (3 субъединицы цитохром-С-оксидазы, 6 компонентов АТФазы и др.), 22 гена транспортных РНК (тРНК) и 2 гена рибосомальных РНК. На рисунке 5-11 представлена схема структуры мтДНК и приведены примеры митохондриальных болезней, которые являются следствием мутации мт-генов. Эти болезни передаются только по материнской линии.
Наследование признаков, передаваемых через ДНК митохондрий, и связь мутаций мтДНК с болезнями человека были впервые показаны в 1988 г. С тех пор обнаружено большое число мутаций мтДНК, лежащих в основе целого ряда нейродегенеративных заболеваний, некоторых МФЗ, митохондриальных миопатий.
Примеры таких заболеваний:
синдром MELAS, развивающийся в связи с мутацией в гене лейциновой тРНК митохондрий и проявляющийся митохондриальной энцефаломиопатией, лактатацидозом и инсультоподобными эпизодами. Вначале развивается сахарный диабет 2 типа с инсулинорезистентностью, который переходит в сахарный диабет 1 типа с инсулинонедостаточностью и наличием аутоантител к антигенам островков поджелудочной железы;
наследственная нейроофтальмия Лебера (характеризуется билатеральной потерей зрения);
синдром MERRF (характеризуется прогрессирующей дегенерацией нервной и мышечной ткани, что проявляется судорогами, атаксией, миопатией, потерей слуха); • летальная инфантильная дыхательная недостаточность и др.
+
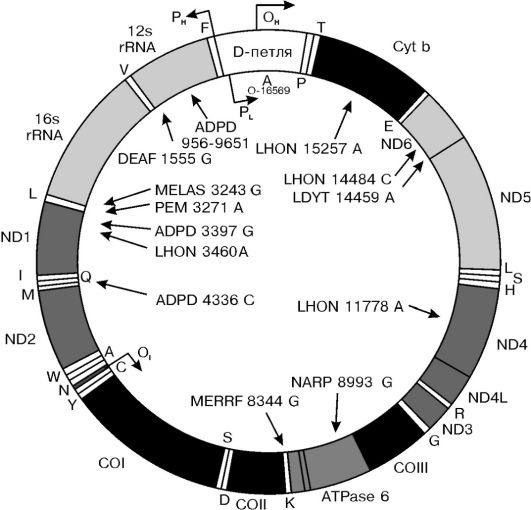 Рис.
5-11. Структура митохондриального генома
и примеры митохондриальных болезней:
APDP - болезнь
Рис.
5-11. Структура митохондриального генома
и примеры митохондриальных болезней:
APDP - болезнь
Альцгеймера/болезнь Паркинсона; DEAF - нейросенсорная потеря слуха; LHON - наследственная нейроофтальмия Лебера; LDYT - LHON и дистония; MELAS - митохондриальная миотония,
энцефалопатия, молочнокислый ацидоз и приступы судорог; MERRF - миоклональная эпилепсия в сочетании с необычно красными мышечными волокнами; NARP - нейропатия, атаксия и пигментный ретинит; PEM - летальная прогрессирующая энцефаломиопатия