

19. Регуляция липидного обмена
Обмен липидов регулируется ЦНС. Центр регуляции находится в промежуточном мозге. Регуляция осуществляется через симпатическую и парасимпатическую систему и через железы внутренней секреции.
Симпатические влияния увеличивают распад и тормозят синтез ТАГ в жировой ткани. Парасимпатические влияния способствуют отложению жира.
Адреналин, норадреналин – усиливают липолиз.
К железам внутренней секреции, через которые нервная система влияет на жировой обмен, относят гипофиз, щитовидную, поджелудочную, половые железы.
Гормон поджелудочной железы – инсулин - регулирует переход углеводов в жиры и стимулирует липогенез: повышает активность фосфатазы, ингибирующую активность ТАГ-липазы и тормозит выход жирных кислот из жировой ткани; Помимо этого инсулин активирует ацетил-КоА-карбоксилазу и синтазу жирных кислот, стимулируя биосинтез ТАГ, холестерина.
Глюкагон стимулирует липолиз, активирует липазу жировой ткани.
Гормоны гипофиза – лютеотропин, соматотропин, вазопрессин, адренокортикотропин усиливают липолиз.
Соматотропный гормон (СТГ) гипофиза стимулирует липолиз.
Гормон щитовидной железы - тироксин активизирует окислительные процессы, стимулирует липолиз.
Глюкокортикоиды в высоких концентрациях оказывают тормозящее влияние на процессы липолиза. В физиологических концентрациях во все тканях, кроме печени, усиливают липолиз.
Гормон лептин, продуцируемый жировыми клетками, предотвращает ожирение.
Мужские половые гормоны – тестостерон – стимулирует липолиз, а женские – липогенез.
20. Абеталипопротеинемия
Абеталипопротеинемия - состояние с аутосомным рецессивным типом наследования обусловлено мутациями в гене микросомного белка-транспортера триглицеридов (ТГ), который необходим для образования хиломикронов и липопротеинов очень низкой плотности (ЛПОНП). В результате жиры пищи не всасываются и сыворотка практически не содержит хиломикронов и ЛПОНП. Абеталипопротеинемию часто впервые обнаруживают у младенцев с нарушением всасывания жиров, стеатореей и отставанием в физическом развитии. Возможна умственная отсталость. Поскольку витамин Е доставляется в периферические ткани ЛПОНП и ЛПНП, в большинстве случаев развивается тяжелый авитаминоз Е. Клинические проявления включают нарушение зрения, обусловленное медленно прогрессирующей дегенерацией сетчатки, сенсорную нейропатию, признаки поражения задних столбов спинного мозга (атаксия и парестезии), а также церебральные симптомы (дисметрию, атаксию и мышечную спастичность), которые могут привести к смерти пациента. Диагноз устанавливают по отсутствию в плазме апопротеина В (Апо В); при исследовании биоптатов кишечника обнаруживается отсутствие микросомального транспортного белка. Отличительной особенностью мазка крови является акантоцитоз эритроцитов. Генетическое тестирование может подтвердить диагноз.
21. Биохимические изменения, приводящие к атеросклерозу
Атеросклероз – это патология, характеризующаяся появлением атерогенных бляшек на внутренней поверхности сосудистой стенки. Одна из основных причин развития такой патологии – нарушение баланса между поступлением холестерола с пищей, его синтезом и выведением из организма. У пациентов, страдающих атеросклерозом, повышены концентрации ЛПНП и ЛПОНП. Существует обратная зависимость между концентрацией ЛПВП и вероятностью развития атеросклероза. Это согласуется с представлениями о функционировании ЛПНП как переносчиков ХС в ткани, а ЛПВП – из тканей.
Базовой метаболической «предпосылкой» развития атеросклероза является гиперхолестеролемия. (повышенное содержание холестерола в крови). Гиперхолестеролемия развивается:
· вследствие избыточного поступления ХС, углеводов и жиров;
· генетической предрасположенности, заключающейся в наследственных дефектах структуры рецепторов ЛПНП или апоВ-100, а также в повышенном синтезе или секреции апоВ-100 (в случае семейной комбинированной гиперлипидемии, при которой в крови повышены концентрации и ХС и ТАГ).
Важную роль в механизмах развития атеросклероза играет модифицирование ЛП. Изменения нормальной структуры липидов и белков в составе ЛПНП делает их чужеродными для организма и поэтому более доступными для захвата фагоцитами. Модифицирование ЛП может происходить по нескольким механизмам:
- гликозилирование белков, происходящее при увеличении концентрации глюкозы в крови;
- перекисная модификация, приводящая к изменениям липидов в липопротеинах и структуры апоВ-100;
- формирование аутоиммунных комплексов ЛП-антитело (изменённые ЛП могут становиться причиной образования аутоантител).
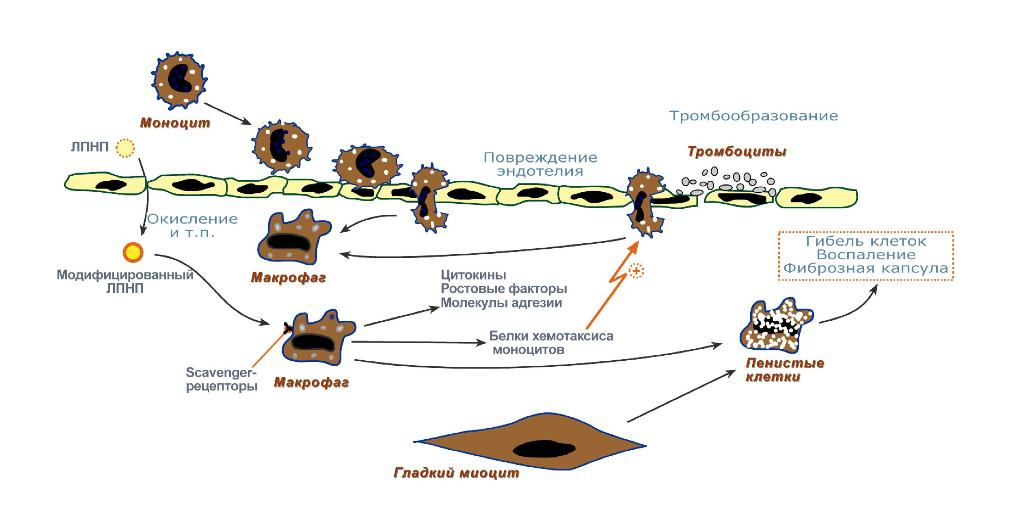
Модифицированные ЛПНП поглощаются макрофагами. Этот процесс не регулируется количеством поглощенного ХС, как в случае его поступления в клетки через специфические рецепторы, поэтому макрофаги перегружаются ХС и превращаются в «пенистые клетки», которые проникают в субэндотелиальное пространство. Это приводит к формированию липидных пятен или полосок в стенке кровеносных сосудов. На этой стадии эндотелий сосудов может сохранять свою структуру.
При увеличении количества пенистых клеток происходит повреждение эндотелия. Повреждение способствует активации тромбоцитов. В результате они секретируют тромбоксан, который стимулирует агрегацию тромбоцитов, а также начинают продуцировать тромбоцитарный фактор роста, стимулирующий пролиферацию гладкомышечных клеток. Последние мигрируют из медиального во внутренний слой артериальной стенки, способствуя таким образом росту бляшки.
Далее происходит прорастание бляшки фиброзной тканью, клетки под фиброзной оболочкой некротизируются, а ХС откладывается в межклеточном пространстве. На последних стадиях развития бляшка пропитывается солями кальция и становится очень плотной. В области бляшки часто образуются тромбы, перекрывающие просвет сосуда, что приводит к острому нарушению кровообращения в соответствующем участке ткани и развитию инфаркта.
22. β-Окисление жирных кислот
Сначала ацил-Коа-синтетазы катализируют образование тиоэфирной связи между карбоксильной группой жирной кислоты и тиоловой группой кофермента А, что приводит к ацил-СоА-производным жирной кислоты и сопряжено с распадом АТР до АМР и РР.
Суммарная реакция: Жирная кислота + CoA + ATP → ацил-CoA +AMP+2Pi
ΔG = - 34 кДж/моль
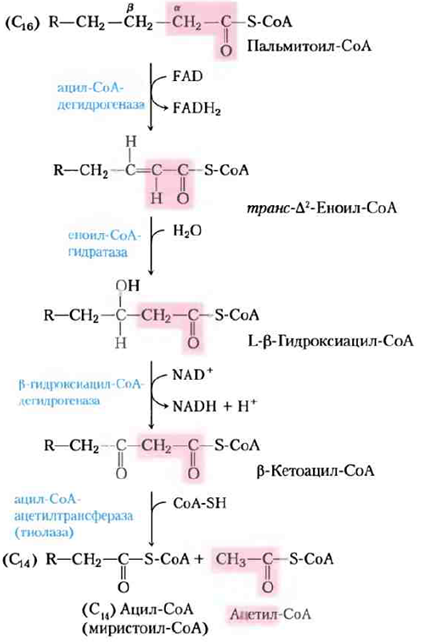
β -Окисление насыщенных жирных кислот проходит в четыре стадии
В первой реакции при дегидрировании ацил-СоА-производного жирной кислоты образуется двойная связь между α- и β -атомами углерода (С-2 и С-3) и получается транс- Δ2-еноил-СоА (символ Δ2 означает положение двойной связи при С-2). Новая двойная связь имеет транс-конфигурацию.
Первая реакция катализируется одним из трех изоферментов, которые называются ацил- СоА-дегидрогеназами; каждый из этих изо-ферментов специфичен к ацилу жирной кислоты с определенной длиной углеродной цепи:
- ацил-СоА-дегидрогеназа очень длинной цепи действует на жирные кислоты с 12-18 атомами углерода
- ацил-СоА-дегидрогеназа средней цепи - на жирные кислоты с 4-14 атомами углерода
- ацил-СоА-дегидрогеназа короткой цепи - на жирные кислоты с 4-8 атомами углерода.
Эти три изофермента относятся к флавопротеинам и содержат FAD в качестве простетической группы. Электроны от ацил-СоА-производного переносятся на FAD, и восстановленная форма дегидрогеназы тут же отдает их переносчику электронов в дыхательной цепи митохондрий - электронпереносящему флавопротеину.
Во второй реакции цикла β-окисления по двойной связи транс- Δ2-еноил- СоА присоединяется вода с образованием L-стереоизомера в-гидроксиацил-CoA (3-гидроксиацил-СоА).
В третьей реакции L- β-гидроксиацил-Соа дегидрируется под действием β-гидроксиацил- CoA-дегидрогеназы, образуя β-кетоацил-Соа; в роли акцептора электронов выступает NAD+. Этот фермент полностью специфичен к L-стереоизомеру гидроксиацил-СоА. Образующийся в реакции NADH отдает электроны NADH- дегидрогеназе, электронному переносчику дыхательной цепи, и, как только электроны попадают на О2, из ADP образуется АТР.
Четвертая (последняя)
реакция цикла β-окисления катализируется
ацил-СоА-ацетилтрансферазой, которую
чаще называют тиолазой и которая
способствует реакции в-кетоацил-СоА с
молекулой свободного кофермента А для
отщепления терминального двухуглеродного
карбоксильного фрагмента исходной
жирной кислоты в виде ацетил-СоА. Другим
продуктом является тиоэфир кофермента
А и жирной кислоты, теперь уже укороченный
на два атома углерода. Эта реакция
называется тиолизом (по аналогии с
гидролизом), поскольку β-кетоацил-Соа
расщепляется в реакции с тиоловой
группой кофермента А.
23. ω-Окисление жирных кислот
Заключается в окислении жирных кислот с ω-атома углерода (то есть самого последнего атома в углеводородной жирнокислотной цепи). Ферменты этого пути у позвоночных локализованы в эндоплазматическом ретикулуме (ЭПР) клеток печени и почек (в отличие от ферментов β-окисления, находящихся в митохондриях). Омега-окислению предпочтительнее подвергаются жирные кислоты с 10—12 углеродными атомами.
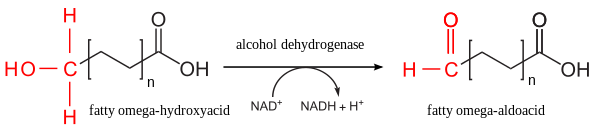
Гидроксилирование. Фермент - оксидаза смешанной функции. В первой реакции на ω-атоме углерода появляется гидроксильная группа. Кислород для этой группы берётся из молекулярного кислорода (О2) в ходе сложной реакции, в которой задействованы цитохром Р450 и NADPH как донор электронов.

Окисление. Фермент - алкогольдегидрогеназа. Гидроксильная группа на ω-атоме окисляется до альдегидной с участием NAD+.
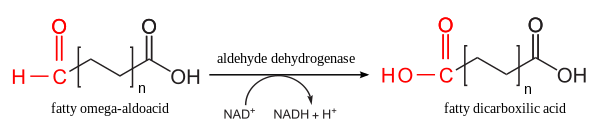
Окисление. Фермент - алкогольдегидрогеназа. Альдегидная группа окисляется до карбоксильной, в результате чего образуется жирная кислота с карбоксильной группой на каждом конце.
После этих трёх реакций каждый из концов может соединиться с коферментом А (СоА), и молекула может поступить в митохондрию и подвергнуться β-окислению. В ходе каждого прохождения β-окисления жирная кислота с двумя концами превращается в дикарбоновую кислоту (например, сукцинат) и адипиновую кислоту. У млекопитающих в норме ω-окисление имеет второстепенное значение, а большая часть жирных кислот разрушается по пути β-окисления. У человека 3,6-диметилоктаноевая кислота и другие разветвлённые жирные кислоты разрушаются преимущественно через ω-окисление. Когда путь β-окисления дефектен (например, из-за мутации или недостатка карнитина), значение ω-окисления увеличивается. ω-окисление также «служит спасением» при дефектном α-окислении (например, у больных синдромом Рефсума). ω-окисление служит эффективным способом элиминирования токсичного повышенного уровня свободных жирных кислот, наблюдающегося при некоторых физиологических состояниях (диабет, голод, алкоголизм)