

- •Foreword
- •Preface
- •Contents
- •1. Introduction to Pathology
- •2. Techniques for the Study of Pathology
- •6. Inflammation and Healing
- •8. Neoplasia
- •16. The Heart
- •17. The Respiratory System
- •18. The Eye, ENT and Neck
- •20. The Gastrointestinal Tract
- •24. The Female Genital Tract
- •25. The Breast
- •26. The Skin
- •27. The Endocrine System
- •28. The Musculoskeletal System
- •29. Soft Tissue Tumours
- •30. The Nervous System
- •Appendix
- •Further Readings
- •Index

871
Chapter 30 |
The Nervous System |
CENTRAL NERVOUS SYSTEM
NORMAL STRUCTURE
The skull and the vertebrae form a rigid compartment encasing the delicate brain and spinal cord. The average weight of the brain is about 1400 gm in men and 1250 gm in women. The two main divisions of the brain— the cerebrum and the cerebellum, are quite distinct in structure. The brain does not have lymphatic drainage. There are 2 types of tissues in the nervous system:
1.Neuroectodermal tissues which include neurons (nerve cells) and neuroglia, and together form the predominant constituent of the CNS.
2.Mesodermal tissues are microglia, dura mater, the leptomeninges (pia-arachnoid), blood vessels and their accompanying mesenchymal cells.
The predominant tissues comprising the nervous system and their general response to injury are briefly considered below:
1. NEURONS. The neurons are highly specialised cells of the body which are incapable of dividing after the first few weeks of birth. Thus, brain damage involving the neurons is irreversible. Neurons vary considerably in size and shape. Their size may range from the small granular cells of the cerebellum to large Betz cells of the motor cortex. Some neurons are round, others oval or fusiform but the prototype of cortical neuron is pyramidal in shape. A neuron consists of 3 main parts: the cell body, an axon and numerous
dendrites (Fig. 30.1,A). The cell body (or perikaryon) is the main constituent of the neuron from which an axon and numerous dendritic processes extend. The cell bodies may be arranged in layers as in the cerebral cortex, or may be aggregated as in the basal ganglia. The cell body possesses a large, round, centrally-placed nucleus having finely granular nuclear chromatin and a prominent nucleolus. The cytoplasm contains polygonal, basophilic structures called Nissl substance. It consists of aggregates of RNA, sheaves of rough endoplasmic reticulum and intervening groups of free ribosomes. Besides Nissl substance, other special features of the cytoplasm of neuronal cell body are the presence of microtubules, synaptic vesicles and neurofilaments which are a form of intermediate filaments specific to neurons. Lipofuscin may be present due to ageing. Neuromelanin is found in neurons in the substantia nigra and pigmented nucleus of the pons.
Neurons respond to injury in a variety of ways depending upon the etiologic agent and the pathologic processes. These include central chromatolysis, atrophy and degeneration of neurons and axons, and intraneuronal storage of substances.
Neuropil is the term used for the fibrillar network formed by processess of all the neuronal cells.
2. NEUROGLIA. The neuroglia provides supportive matrix and maintenance to the neurons. It includes 3 types of cells: astrocytes, oligodendrocytes and ependymal cells (Fig. 30.1,B). Neuroglia is generally referred to as glia; the
System Nervous The 30 CHAPTER
Figure 30.1 
 Cells comprising the nervous system.
Cells comprising the nervous system.

872tumours originating from it are termed gliomas, and reactive proliferation of the astrocytes being called gliosis.
i)Astrocytes. The astrocytes are stellate cells with numerous fine branching processes. In routine haematoxylin and eosin stains, an astrocyte has round or oval vesicular nucleus, but unlike neuron, lacks a prominent nucleolus. The cytoplasm is generally scanty. The processes radiate from the cell body. Depending upon the type of processes, two types of astrocytes are distinguished:
 Protoplasmic astrocytes have branched processes and are found mostly in the grey matter.
Protoplasmic astrocytes have branched processes and are found mostly in the grey matter.
|
Fibrous astrocytes have long, thin processes and are present |
|
|
mainly in the white matter. |
|
|
Some astrocytic processes are directed towards neurons |
|
|
and their processes, which others surround capillaries by |
|
|
terminal expansions called foot processes. The astrocytic |
|
|
processes may not be visible by routine stains but can be |
|
|
demonstrated by phosphotungstic acid haematoxylin |
|
|
(PTAH) stain. Ultrastructurally, these processes are |
|
|
composed of abundant intermediate filaments, mostly |
|
|
vimentin. |
|
SECTION |
The main functions of astrocytes in health are physio- |
|
logical and biochemical support to the neurons and |
||
|
||
|
interactions with capillary endothelial cells to establish blood |
|
|
brain barrier. In case of damage to the brain, astrocytes act |
|
|
like fibroblasts of other tissues. The astrocytes in respond to |
|
III |
injury undergo hyperplasia and hypertrophy termed ‘gliosis’ |
|
which is an equivalent of scar elsewhere in the body. |
||
|
||
|
Gemistocytic astrocytes are early reactive astrocytes having |
|
Systemic |
prominent pink cytoplasm. Long-standing progressive |
|
gliosis results in the development of Rosenthal fibres which |
||
|
||
|
are eosinophilic, elongated or globular bodies present on the |
|
|
astrocytic processes. |
|
Pathology |
Corpora amylacea are basophilic, rounded, sometimes |
|
ii) Oligodendrocytes. Oligodendrocytes are so named |
||
|
laminated bodies, present in elderly people in the white |
|
|
matter and result from accumulation of starch-like material |
|
|
in the degenerating astrocytes. |
|
|
because of their short and fewer processes when examined |
|
|
by light microscopy with special stains (oligo=short). In |
|
|
haematoxylin-eosin stained sections, these cells appear as |
|
|
small cells with a darkly-staining nucleus resembling that of |
|
|
small lymphocyte. The cytoplasm appears as a clear halo |
|
|
around the nucleus. Oligodendrocytes are present |
|
|
throughout the brain in grey as well as white matter and are |
|
|
most numerous of all other cells in the CNS. In grey matter, |
|
|
they are clustered around the neurons and are called satellite |
|
|
cells. In white matter, they are present along the myelinated |
|
|
nerve fibres and are termed interfascicular oligodendroglia. |
|
|
The major function of oligodendrocytes is formation and |
|
|
maintenance of myelin. Thus, in this respect they are |
|
|
counterparts of Schwann cells of the peripheral nervous |
|
|
system. Diseases of oligodendrocytes are, therefore, disorders |
|
|
of myelin and myelinisation such as inherited leucodys- |
|
|
trophies and acquired demyelinating diseases. |
|
|
iii) Ependymal cells. The ependymal cells are epithelium- |
|
|
like and form a single layer of cells lining the ventricular |
system, aqueduct, central canal of the spinal cord and cover the choroid plexus. They are cuboidal to columnar cells and have ciliated luminal surface, just beneath which are present small bodies termed blepharoplasts.
The ependymal cells influence the formation and composition of the cerebrospinal fluid (CSF) by processes of active secretion, diffusion, absorption and exchange. The function of cilia is not very clear but probably they play a role in the circulation of CSF. The ependymal cells respond to injury by cell loss and the space left is filled by proliferation of underlying glial fibres.
3. MICROGLIA. Microglia is the nervous system counterpart of the monocyte-macrophage system. Although the term ‘microglia’ is commonly used but it is inappropriate since these cells, unlike neuroglia, are not of neuroectodermal origin. Microglial cells (or Hortega cells) are not fixed but are mobile cells. These cells are found throughout the brain and are often present close to the blood vessels. Normally, microglial cells appear as small inconspicuous cells with bean-shaped vesicular nuclei, scanty cytoplasm and long cytoplasmic processes (Fig. 30.1,C). In response to injury or damage, however, these cells have capability to enlarge in size, proliferate and develop elongated nuclei, so called rod cells. Microglial cells may actually assume the shape and phagocytic function of macrophages and form gitter cells. The foci of necrosis and areas of selective hypoxic damage to the neurons are surrounded by microglial cells which perform phagocytosis of damaged and necrosed cells; this is known
as neuronophagia.
4.DURA MATER. The dura mater is a tough fibrous covering of the brain which is closely attached to the skull on its inner layer of endocranial periosteum. In the region of spinal canal, it encloses a potential space, the epidural space, between the bone and the dura. The dura is composed of dense collagen, fused with periosteum of the skull.
5.PIA-ARACHNOID (LEPTOMENINGES). The leptomeninges (lepto=thin, slender) consisting of the pia and arachnoid mater form the delicate vascular membranous covering of the central nervous system. The pia mater is closely applied to the brain and its convolutions, while the arachnoid mater lies between the pia mater and the dura mater without dipping into sulci. Thus, a space is left between the two layers of leptomeninges, known as subarachnoid space, which contains the CSF. The major arteries and veins run in the subarachnoid space and small nutrient arteries pass into the cortex. Extension of the subarachnoid space between the wall of blood vessels entering the brain and their pial sheaths form a circumvascular space called Virchow-Robin space. Another important potential space is enclosed between the dura and the arachnoid membrane known as subdural space.
DEVELOPMENTAL ANOMALIES
These malformations are the result of various inherited and acquired factors. The acquired conditions include viral infections of the mother and foetus (e.g. rubella), intake of drugs (e.g. thalidomide), exposure to ionising radiation and foetal anoxia. There are a large number of developmental

malformations of the CNS but only a few important and common ones are mentioned here. Congenital hydrocephalus is considered separately along with other types of hydrocephalus.
Spinal Cord Defects
Spina bifida is the term applied to the malformations of the vertebral column involving incomplete embryologic closure of one or more of the vertebral arches (rachischisis), most frequently in the lumbosacral region. The vertebral defect is frequently associated with defect in the neural tube structures and their coverings. The bony defect may be of varying degree. The least serious form is spina bifida occulta in which there is only vertebral defect but no abnormality of the spinal cord and its meninges. The site of bony defect is marked by a small dimple, or a hairy pigment mole in the overlying skin. The larger bony defect, however, appears as a distinct cystic swelling over the affected site called spina bifida cystica. This is associated with herniation of the meninges or the spinal cord, or both.

 Herniation of the meninges alone through the bony defect, meningocele, is a less common variety. The herniated sac in meningocele consists of dura and arachnoid.
Herniation of the meninges alone through the bony defect, meningocele, is a less common variety. The herniated sac in meningocele consists of dura and arachnoid.
 The commonest and more serious form is, however, meningomyelocele in which the spinal cord or its roots also herniate through the defect and are attached to the posterior wall of the sac. In this defect, the dura and the skin in the sac are deficient. A more serious variant of meningomyelocele is associated with hydrocephalus and Arnold-Chiari malformation.
The commonest and more serious form is, however, meningomyelocele in which the spinal cord or its roots also herniate through the defect and are attached to the posterior wall of the sac. In this defect, the dura and the skin in the sac are deficient. A more serious variant of meningomyelocele is associated with hydrocephalus and Arnold-Chiari malformation.

 A rare form of the defect is myelocele or syringomyelocele in which there is defective closure of the spinal canal so that the sac consists of an open flat neural tissue plate without skin covering and the CSF leaking through it.
A rare form of the defect is myelocele or syringomyelocele in which there is defective closure of the spinal canal so that the sac consists of an open flat neural tissue plate without skin covering and the CSF leaking through it.
Meningomyelocele and myelocele are frequently associated with neurologic defects of varying degree which include bladder and bowel dysfunction, motor and sensory defects, and paraplegia.
The existence of defect in bony closure in the region of occipital bone or fronto-ethmoid junction may result in cranial meningocele and encephalocele.
Syringomyelia and Syringobulbia
These are congenital malformations which manifest clinically later in life and often develop in association with certain acquired lesions involving the CNS. Syringomyelia and syringobulbia are characterised by development of a syrinx or a tubular cavity in the spinal cord and medulla respectively. The cavity may be fusiform or irregular. It usually begins in the grey matter of the spinal cord dorsal to the central canal. The syrinx is usually surrounded by glial tissue. If the cavity communicates with the spinal canal, it is lined by ependymal cells. Since the fibres of lateral spinothalamic tract are frequently involved in the cavity, the clinical effects include loss of pain and temperature sensation in the affected region.
Arnold-Chiari Malformations
Arnold-Chiari malformation is the term used for a group of malformations of the brain involving the brainstem and cerebellum. The primary defect is elongation of the medulla and part of the vermis of the cerebellum resulting from failure of the pontine flexure to form. Approximately 50% of children with hydrocephalus have the Arnold-Chiari malformation. Four types are described, of which type II malformation is the most common and is most frequently associated with congenital hydrocephalus. Most patients of Arnold-Chiari malformation have, in addition, meningomyelocele. The major components of type II Arnold-Chiari malformation are as follows:
1.Elongation of the medulla with part of fourth ventricle in the cervical canal.
2.Distortion of the medulla forming a characteristic S- shaped bend at the junction with the cervical spinal cord.
3.Lengthening and herniation of the cerebellar vermis and cerebellar tonsils through the foramen magnum resulting in formation of a mass over the upper cervical cord.
Combination of these abnormalities results in stenosis of the aqueduct or obstruction of the foramina of Luschka and Magendie causing internal hydrocephalus (discussed below).
HYDROCEPHALUS
Hydrocephalus is the term used for increased volume of CSF within the skull, accompanied by dilatation of the ventricles. In majority of cases of hydrocephalus, there is increased intracranial pressure. This type of hydrocephalus involving ventricular dilatation is termed internal hydrocephalus. A localised collection of CSF in the subarachnoid space is called external hydrocephalus. For better understanding of causes and mechanisms involved in the hydrocephalus, it is essential to briefly review the source and circulation of CSF.
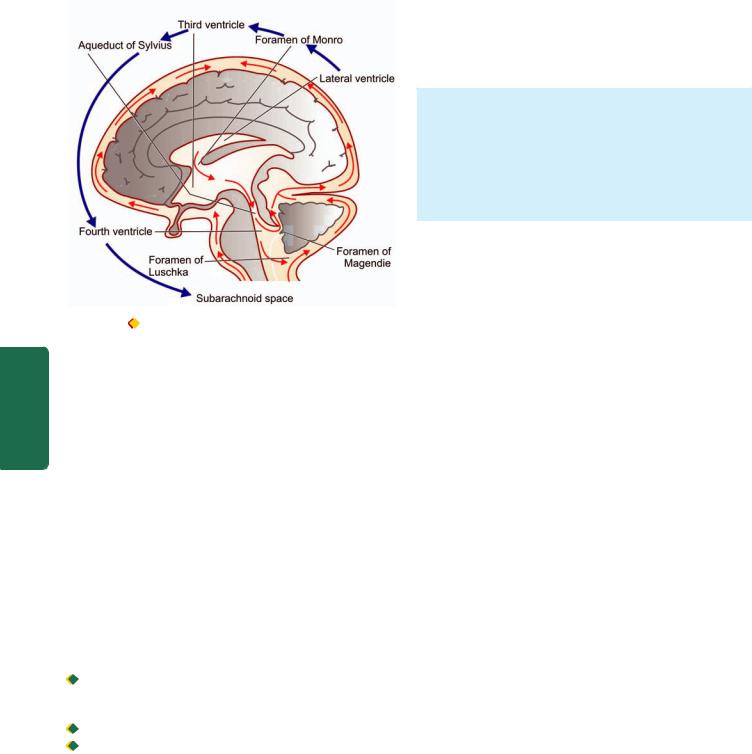
Source and Circulation of CSF
CSF is mainly produced by choroid plexus in the lateral, third and fourth ventricle, and a small part is formed on the surface of the brain and spinal cord. The total volume of CSF is about 120-150 ml. CSF formed in the lateral ventricles flows through the foramina of Munro to the third ventricle and from there by the aqueduct of Sylvius to the fourth ventricle. The fluid then passes through the foramina of Magendie and Luschka of the fourth ventricle to reach the subarachnoid space of the brain. It then spreads through the subarachnoid space over the surface of the spinal cord. It is absorbed into the blood by the arachnoid villi present along the dural venous sinuses
(Fig. 30.2).
Types and Etiopathogenesis
Hydrocephalus is classified into primary and secondary types, the former being much more common, both types have distinct etiology and pathogenesis.
PRIMARY HYDROCEPHALUS. Primary hydrocephalus is defined as actual increase in the volume of CSF within the skull along with elevated intracranial pressure. There are 3 possible mechanisms of primary hydrocephalus:
873
System Nervous The 30 CHAPTER
874
|
Figure 30.2 |
Normal circulation of CSF. |
||
SECTION |
1. |
Obstruction to the flow of CSF. |
||
commonest cause and is termed obstructive hydrocephalus. The |
||||
|
2. |
Overproduction of CSF. |
||
|
3. |
Deficient reabsorption of CSF. |
||
|
|
However, obstruction to the flow of CSF is by far the |
||
III |
terms non-communicating and communicating hydrocephalus |
|||
are used to denote the site of obstruction: |
||||
|
||||
Systemic |
Non-communicating hydrocephalus. When the site of |
|||
obstruction of CSF pathway is in the third ventricle or at the |
||||
|
||||
|
exit foramina in the fourth ventricle, the ventricular system |
|||
|
enlarges and CSF cannot pass into the subarachnoid space. |
|||
|
This is termed as non-communicating hydrocephalus. |
|||
Pathology |
Among the common causes are the following: |
|||
ii) |
Acquired non-communicating hydrocephalus may occur from |
|||
|
i) |
Congenital non-communicating hydrocephalus e.g. stenosis |
||
|
of the aqueduct, Arnold-Chiari malformation, progressive |
|||
|
gliosis of the aqueduct and intra-uterine meningitis. |
|||
|
expanding lesion within the skull. These conditions are as |
|||
|
under: |
|
||
|
|
Tumours adjacent to the ventricular system e.g. |
||
|
|
ependymoma, choroid plexus papilloma, medullo- |
||
|
|
blastoma and others. |
||
|
|
Inflammatory lesions e.g. cerebral abscess, meningitis. |
||
|
|
Haemorrhage e.g. parenchymal haemorrhage, intra- |
||
|
|
ventricular haemorrhage, and epidural and subdural |
||
|
|
haematoma. |
||
Communicating hydrocephalus. When obstruction to the flow of CSF is in the subarachnoid space at the base of the brain, it results in enlargement of the ventricular system but CSF flows freely between dilated ventricles and the spinal canal. This is called communicating hydrocephalus. The causes of communicating hydrocephalus are non-obstructive which are as follows:
i)Overproduction of CSF e.g. choroid plexus papilloma.
ii)Deficient reabsorption of CSF e.g. following meningitis, subarachnoid haemorrhage and dural sinus thrombosis.
SECONDARY HYDROCEPHALUS. Secondary hydrocephalus is much less common and is defined as compensatory increase of CSF due to loss of neural tissue without associated rise in intracranial pressure (normal pressure hydrocephalus) e.g. from cerebral atrophy and infarction.
MORPHOLOGIC FEATURES. Grossly, there is dilatation of the ventricles depending upon the site of obstruction. There is thinning and stretching of the brain. The scalp veins overlying the enlarged head are engorged and the fontanelle remain open.
Histologically, severe hydrocephalus may be associated with damage to ependymal lining of the ventricles and periventricular interstitial oedema.
INFECTIONS
A large number of pathogens comprising various kinds of bacteria, fungi, viruses, rickettsiae and parasites can cause infections of the nervous system. The micro-organisms may gain entry into the nervous system by one of the following routes:
1.Via blood stream. Spread of infection by the arterial route from another focus is the most common mode of spread of infection in the nervous system. Less often, the spread may occur by retrograde venous route and by lodgement of septic emboli in the brain.
2.Direct implantation. Spread of infection by direct implantation occurs following skull fractures or through defects in the bony and meningeal coverings of the nervous system.
3.Local extension. Extension of infection from contiguous focus such as otitis media and frontal or mastoid sinusitis may occur.
4.Along nerve. Certain viruses such as herpes simplex, herpes zoster and rabies spread along cranial and peripheral nerves and ascend to the CNS.
In general, resultant lesions are in the form of either diffuse inflammation of the meninges (meningitis) and of brain parenchyma (encephalitis), or combination of both (meningoencephalitis). In addition, other inflammatory lesions of CNS include: brain abscess, epidural abscess, subdural empyema, septic thromboembolism of dural sinuses and encephalomyelitis. Some of the morphologically significant lesions are described below.
MENINGITIS
Meningitis is inflammatory involvement of the meninges. Meningitis may involve the dura called pachymeningitis, or the leptomeninges (pia-arachnoid) termed leptomeningitis. The latter is far more common, and unless otherwise specified, meningitis would mean leptomeningitis.
Pachymeningitis is invariably an extension of the inflammation from chronic suppurative otitis media or from fracture of the skull. An extradural abscess may form by suppuration between the bone and dura. Further spread of infection may penetrate the dura and form a subdural abscess. Other effects of pachymeningitis are localised or generalised leptomeningitis and cerebral abscess.
Leptomeningitis, commonly called meningitis, is usually the result of infection but infrequently chemical meningitis and carcinomatous meningitis by infiltration of the subarachnoid space by cancer cells may occur. Infectious meningitis is broadly classified into 3 types: acute pyogenic, acute lymphocytic (viral, aseptic) and chronic (bacterial or fungal).
Acute Pyogenic Meningitis
Acute pyogenic or acute purulent meningitis is acute infection of the pia-arachnoid and of the CSF enclosed in the subarachnoid space. Since the subarachnoid space is continuous around the brain, spinal cord and the optic nerves, infection spreads immediately to whole of the cerebrospinal meninges as well as to the ventricles.
ETIOPATHOGENESIS. The causative organisms vary with age of the patient:
1.Escherichia coli infection is common in neonates with neural tube defects.
2.Haemophilus influenzae is commonly responsible for infection in infants and children.
3.Neisseria meningitidis causes meningitis in adolescent and young adults and is causative for epidemic meningitis.
4.Streptococcus pneumoniae is causative for infection at extremes of age and following trauma.
The routes of infection in acute pyogenic meningitis are as follows:
1.Most commonly by the blood stream.
2.From an adjacent focus of infection.
3.By iatrogenic infection such as introduction of microorganisms at operation or during lumbar puncture.
MORPHOLOGIC FEATURES. Grossly, pus accumulates in the subarachnoid space so that normally clear CSF becomes turbid or frankly purulent. The turbid fluid is particularly seen in the sulci and at the base of the brain where the space is wide. In fulminant cases, some degree of ventriculitis is also present having a fibrinous coating on their walls and containing turbid CSF. In addition, purulent material may interfere with CSF flow and result in obstructive hydrocephalus.
Microscopically, there is presence of numerous polymorphonuclear neutrophils in the subarachnoid space as well as in the meninges, particularly around the blood vessels (Fig. 30.3). Gram-staining reveals varying number of causative bacteria.
CLINICAL FEATURES AND DIAGNOSIS. Acute bacterial meningitis is a medical emergency. The immediate clinical manifestations are fever, severe headache, vomiting, drowsiness, stupor, coma, and occasionally, convulsions. The most important clinical sign is stiffness of the neck on forward bending.
The diagnosis is confirmed by examining CSF as soon as possible. The diagnostic alterations in the CSF in acute pyogenic meningitis are as under:
1. Naked eye appearance of cloudy or frankly purulent CSF.
875
Figure 30.3 |
Acute suppurative meningitis. |
2. Elevated CSF pressure (above 180 mm water). |
CHAPTER |
||
|
|||
3. Polymorphonuclear neutrophilic leucocytosis in CSF |
|
||
(between 10-10,000/μl). |
|
||
4. Raised CSF protein level (higher than 50 mg/dl). |
|
||
5. Decreased CSF sugar concentration (lower than 40 mg/ |
30 |
||
dl). |
|||
|
|||
6. Bacteriologic examination by Gram’s stain or by CSF |
|
||
culture reveals causative organism. |
The |
||
Acute Lymphocytic (Viral, Aseptic) Meningitis |
|||
Nervous |
|||
Acute lymphocytic meningitis is a viral or aseptic menin- |
|||
|
|||
gitis, especially common in children and young adults. |
|
||
Among the etiologic agents are numerous viruses such as |
System |
||
enteroviruses, mumps, ECHO viruses, coxsackie virus, |
|||
|
|||
Epstein-Barr virus, herpes simplex virus-2, arthropode-borne |
|
||
viruses and HIV. However, evidence of viral infection may |
|
||
not be demonstrable in about a third of cases. |
|
||
|
|
|
|
MORPHOLOGIC FEATURES. Grossly, some cases show |
|
|
|
swelling of the brain while others show no distinctive |
|
|
|
change. |
|
|
|
Microscopically, there is mild lymphocytic infiltrate in |
|
|
|
the leptomeninges. |
|
|
|
CLINICAL FEATURES AND DIAGNOSIS. The clinical |
|
||
manifestations of viral meningitis are much the same as in |
|
||
bacterial meningitis with features of acute onset meningeal |
|
||
symptoms and fever. However, viral meningitis has a benign |
|
||
and self-limiting clinical course of short duration and is |
|
||
invariably followed by complete recovery without the life- |
|
||
threatening complications of bacterial meningitis. |
|
||
The CSF findings in viral meningitis are as under: |
|
||
1.Naked eye appearance of clear or slightly turbid CSF.
2.CSF pressure increased (above 250 mm water).
3.Lymphocytosis in CSF (10-100 cells/μl).
8764. CSF protein usually normal or mildly raised.
5.CSF sugar concentration usually normal.
6.CSF bacteriologically sterile.
Chronic (Tuberculous and Cryptococcal) Meningitis
There are two principal types of chronic meningitis—one bacterial (tuberculous meningitis) and the other fungal (cryptococcal meningitis). Both types cause chronic granulomatous reaction and may produce parenchymal lesions.
 Tuberculous meningitis occurs in children and adults through haematogenous spread of infection from tuberculosis elsewhere in the body, or it may simply be a manifestation of miliary tuberculosis. Less commonly, the spread may occur directly from tuberculosis of a vertebral body.
Tuberculous meningitis occurs in children and adults through haematogenous spread of infection from tuberculosis elsewhere in the body, or it may simply be a manifestation of miliary tuberculosis. Less commonly, the spread may occur directly from tuberculosis of a vertebral body.
 Cryptococcal meningitis develops particularly in debilitated or immunocompromised persons, usually as a result of haematogenous dissemination from a pulmonary lesion. Cryptococcal meningitis is especially an important cause of
Cryptococcal meningitis develops particularly in debilitated or immunocompromised persons, usually as a result of haematogenous dissemination from a pulmonary lesion. Cryptococcal meningitis is especially an important cause of
|
meningitis in patients with AIDS. |
|
SECTION |
|
|
MORPHOLOGIC FEATURES. Grossly, in tuberculous |
||
meningitis, the subarachnoid space contains thick exudate, |
||
particularly abundant in the sulci and the base of the brain. |
||
Tubercles, 1-2 mm in diameter, may be visible, especially |
||
adjacent to the blood vessels. The exudate in cryptococcal |
||
III |
meningitis is scanty, translucent and gelatinous. |
|
Microscopically, tuberculous meningitis shows exudate |
||
|
||
Systemic |
of acute and chronic inflammatory cells, and granulomas |
|
with or without caseation necrosis and giant cells. Acid- |
||
fast bacilli may be demonstrated. Late cases show dense |
||
fibrous adhesions in the subarachnoid space and |
||
consequent hydrocephalus. Cryptococcal meningitis is |
||
Pathology |
characterised by infiltration by lymphocytes, plasma cells, |
|
CLINICAL FEATURES AND DIAGNOSIS. Tuberculous |
||
|
an occasional granuloma and abundant characteristic |
|
|
capsulated cryptococci. |
|
|
meningitis manifests clinically as headache, confusion, |
Figure 30.4 
 Cryptococci in CSF as seen in mucicarmine stain.
Cryptococci in CSF as seen in mucicarmine stain.
malaise and vomiting. The clinical course in cryptococcal meningitis may, however, be fulminant and fatal in a few weeks, or be indolent for months to years.
The CSF findings in chronic meningitis are as under:
1.Naked eye appearance of a clear or slightly turbid CSF which may form fibrin web on standing.
2.Raised CSF pressure (above 300 mm water).
3.Mononuclear leucocytosis consisting mostly of lymphocytes and some macrophages (100-1000 cells/μl).
4.Raised protein content.
5.Lowered glucose concentration.
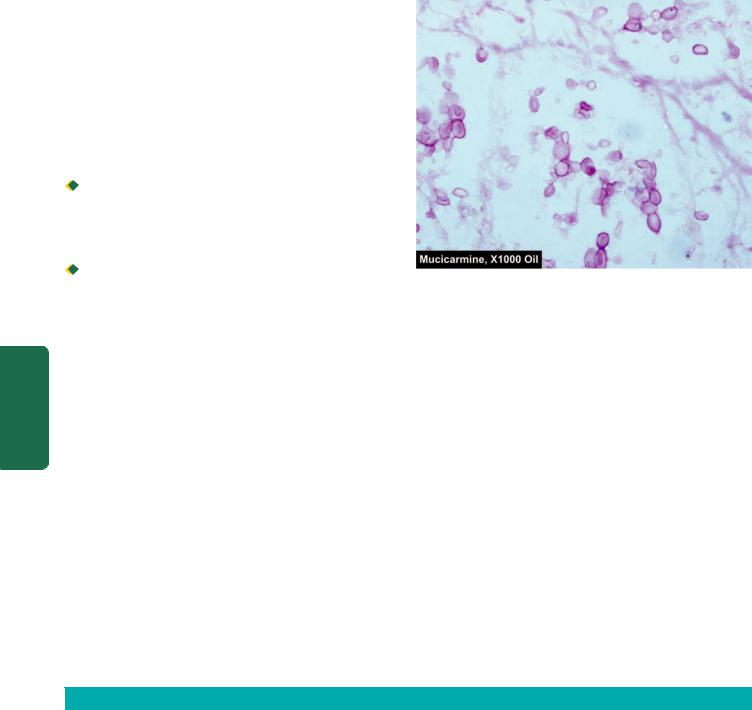
6.Tubercle bacilli may be found on microscopy of centrifuged deposits by ZN staining in tuberculous meningitis. Pathognomonic capsulated cryptococci with a halo are appreciated in India ink preparation of CSF in cases of cryptococcal meningitis, while the capsule is better demonstrated by mucicarmine stain (Fig. 30.4).
Table 30.1 summarises the CSF findings in the three important types of meningitis in comparison with those in health.
TABLE 30.1: CSF Findings in Health and Various Types of Meningitis.
|
Feature |
Normal |
Acute Pyogenic |
Acute Lympho- |
Chronic (Tuberculous) |
|
|
|
(Bacterial) |
cytic (Viral) |
Meningitis |
|
|
|
Meningitis |
Meningitis |
|
1. |
Naked eye |
Clear and colourless |
Cloudy or frankly |
Clear or slightly |
Clear or slightly turbid, forms |
|
appearance |
|
purulent |
turbid |
fibrin coagulum on standing |
2. |
CSF pressure |
60-150 mm water |
Elevated |
Elevated |
Elevated |
|
|
|
(above 180 mm water) |
(above 250 mm water) |
(above 300 mm water) |
3. |
Cells |
0-4 |
10-10,000 |
10-100 |
100-1000 |
|
|
lymphocytes/μl |
neutrophils/μl |
lymphocytes/μl |
lymphocytes/μl |
4. |
Proteins |
15-45 mg/dl |
Markedly raised |
Raised |
Raised |
5. |
Glucose |
50-80 mg/dl |
Markedly reduced |
Normal |
Reduced |
6. |
Bacteriology |
Sterile |
Causative |
Sterile |
Tubercle bacilli |
|
|
|
organisms present |
|
present |
|
|
|
|
|
|
ENCEPHALITIS
Parenchymal infection of brain is termed encephalitis. Encephalitis may be the result of bacterial, viral, fungal and protozoal infections.
Bacterial Encephalitis
Bacterial infection of the brain substance is usually secondary to involvement of the meninges rather than a primary bacterial parenchymal infection. This results in bacterial cerebritis that progresses to form brain abscess. However, tuberculosis and neurosyphilis are the two primary bacterial involvements of the brain parenchyma.
BRAIN ABSCESS. Brain abscesses may arise by one of the following routes:
1.By direct implantation of organisms e.g. following compound fractures of the skull.
2.By local extension of infection e.g. chronic suppurative otitis media, mastoiditis and sinusitis.
3.By haematogenous spread e.g. from primary infection in the heart such as acute bacterial endocarditis, and from lungs such as in bronchiectasis.
Clinically, there is usually evidence of reactivation of infection at the primary site preceding the onset of cerebral symptoms. The features of abscess are fever, headache, vomiting, seizures and focal neurological deficits depending upon the location of the abscess. Brain abscess is most common in cerebral hemispheres and less frequent in the cerebellum and basal ganglia.
Grossly, it appears as a localised area of inflammatory necrosis and oedema surrounded by fibrous capsule. Microscopically, the changes consist of liquefactive necrosis in the centre of the abscess containing pus. It is surrounded by acute and chronic inflammatory cells, neovascularisation, oedema, septic thrombosis of vessels, fibrous encapsulation and zone of gliosis. The CSF and overlying meninges also show evidence of acute and chronic inflammation.
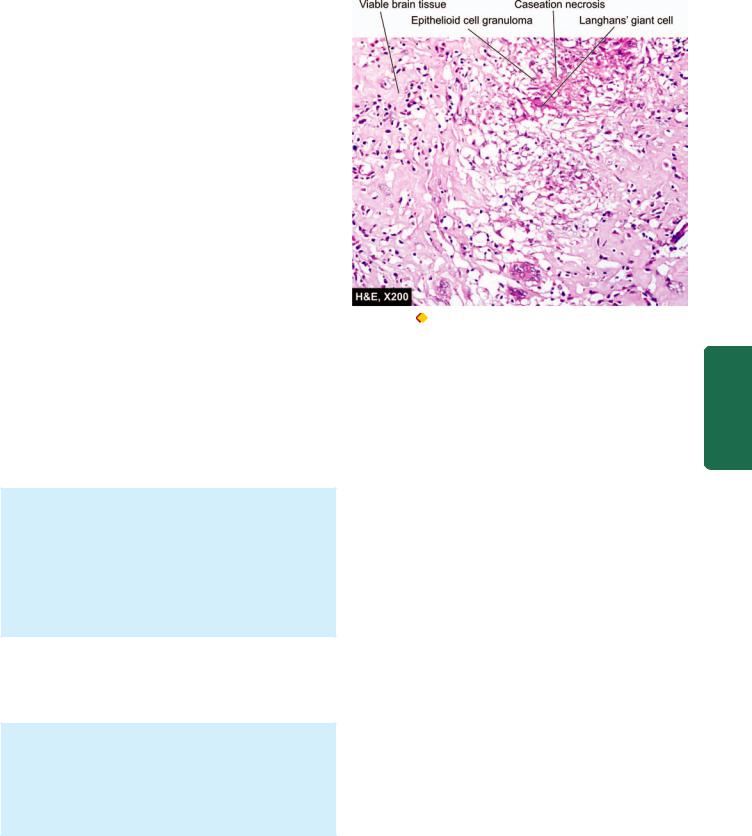
TUBERCULOMA. Tuberculoma is an intracranial mass occurring secondary to dissemination of tuberculosis elsewhere in the body. Tuberculomas may be solitary or multiple.
Grossly, it has a central area of caseation necrosis surrounded by fibrous capsule.
Microscopically, there is typical tuberculous granulomatous reaction around the central caseation necrosis (Fig. 30.5). A zone of gliosis generally surrounds the tuberculoma. Advanced cases may show areas of calcification.
NEUROSYPHILIS. Syphilitic lesions of the CNS used to be common and serious, but more recently there is evidence of atypical neurosyphilis in cases of HIV/AIDS. The lesions in syphilis may be in the form of syphilitic meningitis found in secondary syphilis, and neurosyphilis consisting of tabes
877
Figure 30.5 |
Tuberculous encephalitis of the brain. |
|
|
dorsalis and generalised paralysis of the insane occurring in |
CHAPTER |
||
tertiary stage (page 161). |
|||
Syphilitic meningitis. This is a form of chronic meningitis |
|||
|
|||
characterised by distinctive perivascular inflammatory |
|
||
reaction of plasma cells and endarteritis obliterans. |
|
||
Tabes dorsalis (Locomotor ataxia). There is slowly pro- |
30 |
||
gressive degeneration of the posterior roots of the spinal |
|||
nerves and the posterior columns of the spinal cord by the |
|
||
spirochaetes. These changes produce loss of coordination of |
The |
||
muscles and joints resulting in locomotor ataxia. These |
|||
|
|||
changes produce loss of coordination of muscles and joints |
Nervous |
||
resulting in locomotor ataxia. There is also loss of pain |
|||
|
|||
sensation and presence of Argyll-Robertson pupils which |
|
||
react to accommodation but not to light. |
|
||
General paralysis of the insane. This is the result of diffuse |
System |
||
parenchymal involvement by the spirochaetes with widespread lesions in the nervous system. The symptoms consist of motor, sensory and psychiatric abnormalities.
Viral Encephalitis
A number of viruses can infect the CNS and produce either aseptic meningitis (described already) or viral encephalitis, but sometimes combination of both termed meningoencephalitis, is present. Most viral infections of the CNS are the end-result of preceding infection in other tissues and organs. There is usually a preceding phase of extraneural viral replication before involvement of the nervous system occurs.
Most of the viruses reach the nervous system via blood stream before which they enter the body by various routes e.g. infection of the skin and mucous membrane (in herpes simplex and herpes zoster-varicella), by the alimentary tract (in enteroviruses including polio virus), by arthropod bite (in arbovirus), by transplacental infection (in cytomegalovirus), and through body fluids in AIDS (in HIV infection).

878Rabies virus travels along the peripheral nerves to reach the CNS. Herpes zoster-varicella is a distinct primary disease (chickenpox) but the virus remains latent for a long time before it gets reactivated to cause severe hyperalgesia and pain along the distribution of nerve related to acutely inflamed posterior root ganglia (herpes zoster). All these viral infections enumerated so far cause acute viral encephalitis. Slow virus diseases are another group of CNS infections in which the agents have not only a long latent period but the disease also develops slowly and may produce subacute sclerosing panencephalitis, progressive multifocal leucoencephalopathy, progressive rubella panencephalopathy and subacute spongiform encephalopathy.
|
MORPHOLOGIC FEATURES. Although histologic |
||
|
changes vary from one viral infection of the CNS to the |
||
|
other but, in general, the characteristic features of viral |
||
|
diseases of the CNS are as under: |
||
|
1. |
Parenchymal infiltrate, chiefly in perivascular location, |
|
|
of mononuclear cells consisting of lymphocytes, plasma |
||
|
cells and macrophages. |
||
|
2. |
Microscopic clusters of microglial cells and presence |
|
SECTION |
of neuronophagia. |
||
3. |
Intranuclear inclusion bodies in most viral diseases |
||
|
|||
|
and specific cytoplasmic inclusions of Negri bodies in |
||
|
rabies. |
||
|
|
|
|
III |
HIV Encephalopathy (AIDS-Dementia Complex) |
|
Next to knocking off of the immune system, HIV has profound |
||
|
||
Systemic |
neurovirulence but unlike tropism for CD4+ T cells of the |
|
microglial cells. HIV infection then sets in a cascade of toxic |
||
|
immune system, HIV does not have neurotropism. HIV has |
|
|
not been identified to infect the neuronal cells but instead |
|
|
infects the cells of monocyte-macrophage cell line including |
|
Pathology |
mediators and cytokines—TNF-α,IL-1, IL-6, TGF-β, |
|
are listed in Table 30.2. Late in the course of AIDS, a group |
||
|
IFN-γ, platelet activating factor (PAF) and endothelin, all of |
|
|
which cause damage to the neuroglial tissues. |
|
|
Important forms of CNS diseases in patients with AIDS |
|
|
of signs and symptoms of CNS disease appear termed HIV |
|
|
encephalopathy or AIDS-dementia complex. One major |
|
|
clinical feature of this entity is the occurrence of dementia i.e. |
|
|
fall in the cognitive ability of the individual compared to |
|
|
previous level. The condition is believed to be the result of |
|
|
direct effect of HIV on the CNS. Clinically, the disease |
|
|
develops in about 25% cases of AIDS while autopsy studies |
|
|
reveal presence of HIV-encephalopathy in 80-90% cases of |
|
|
AIDS. |
|
|
|
|
|
Histologically, the changes are more in subcortical area |
|
|
of the brain and consist of gliosis, multinucleate giant cell |
|
|
encephalitis, and vacuolar myelopathy. |
|
|
Progressive Multifocal Leucoencephalopathy |
|
|
Progressive multifocal leucoencephalopathy (PML) is a slow |
|
|
viral infection of the CNS caused by a papovavirus called JC |
|
|
virus (not to be confused with CJ disease or mad-cow disease; |
|
|
JC virus here stands for the initials of the patient first |
TABLE 30.2. Major Forms of CNS Diseases in AIDS.
|
Disease |
Incidence |
|
|
|
|
|
1. |
HIV-encephalopathy (AIDS-dementia complex) |
25% |
|
2. |
Opportunistic infections |
|
|
|
i) |
Toxoplasmosis |
15% |
|
ii) |
Cryptococcal meningitis |
9% |
|
iii) |
Progressive multifocal leucoencephalopathy |
4% |
|
iv) |
Neurosyphilis |
1% |
|
v) |
Tuberculous meningitis |
1% |
3. |
Neoplasms |
|
|
|
i) |
Primary CNS lymphoma |
4% |
|
ii) |
Kaposi’s sarcoma |
<1% |
4. |
Progressive multifocal leucoencephalopathy (PML) |
1% |
|
5. |
Peripheral neuropathies |
70% |
|
6. |
Myelopathy (Spinal cord disease) |
20% |
|
|
|
|
|
infected). PML develops in immunocompromised individual like CMV and Toxoplasma encephalitis does, and is, therefore, an important form of encephalitis due to increasing number of cases of AIDS.
PML infects oligodendrocytes and causes progressive demyelination at multifocal areas scattered throughout the CNS.
Grossly, the lesions consist of focal, irregular gelatinous areas most prominent at the junction of grey and white matter. Main areas affected are cerebrum, brainstem, cerebellum, and sometimes spinal cord.
Microscopically, the features are as under:  Focal areas of demyelination.
Focal areas of demyelination.
 Many lipid-laden macrophages in the centre of foci.
Many lipid-laden macrophages in the centre of foci.  Enlarged oligodendroglial nuclei containing purple
Enlarged oligodendroglial nuclei containing purple
viral inclusions at the periphery of the lesion.
Spongiform Encephalopathy
(Creutzfeldt-Jakob Disease)
Spongiform encephalopathy, also called Creutzfeldt-Jakob disease (CJD) or mad-cow disease, though included under the group of viral encephalitis but is caused by accumulation of prion proteins. Prion proteins are a modified form of normal structural proteins present in the mammalian CNS and are peculiar in two respects: they lack nucleic acid (DNA or RNA), and they can be transmitted as an infectious proteinaceous particles (Dr Prusiner was awarded the Nobel Prize in medicine in 1997 for his discovery on prion proteins).
Majority of cases occur sporadically though familial predisposition with autosomal dominant inheritance has also been reported in 5-15% cases. Other methods of transmission are by iatrogenic route (e.g. by tissue transplantation from an infected individual) and by human consumption of BSE (bovine spongiform encephalopathy)-infected beef, also called as mad-cow disease.
Clinically, CJD is characterised by rapidly progressive dementia with prominent association of myoclonus. CJD is invariably fatal with mean survival of about 7 months after diagnosis.
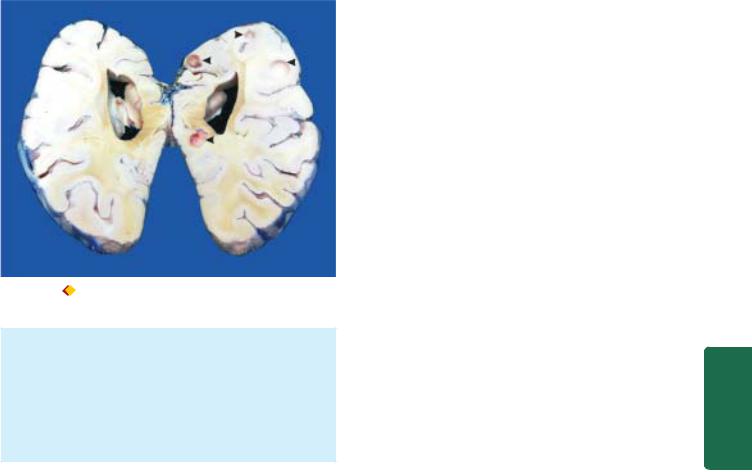
Figure 30.6 
 Neurocysticercosis. The sliced surface of the cerebral hemisphere of the brain shows may tiny whitish nodules and cysts about 1 cm in diameter.
Neurocysticercosis. The sliced surface of the cerebral hemisphere of the brain shows may tiny whitish nodules and cysts about 1 cm in diameter.
Grossly, the changes are too rapid to become noticeable but brain atrophy may be seen in long-standing cases. Microscopically, the hallmark is spongiform change i.e. there are small round vacuoles in the neuronal cells. These changes are predominantly seen in the cortex and other grey matter areas. Spongiform changes result in neuronal loss and glial cell proliferation but significantly without any inflammation or white matter involvement.
Fungal and Protozoal Encephalitis
Mycotic diseases of the CNS usually develop by blood stream from systemic deep mycoses elsewhere in the body. They are particularly more common in immunosuppressed individuals such as in AIDS, patients of lymphomas and other cancers. Some of the fungi which may disseminate to
the CNS are Candida albicans, Mucor, Aspergillus fumigatus,
Cryptococcus neoformans, Histoplasma capsulatum and Blastomyces dermatitidis. These fungal infections may produce one of the three patterns: fungal chronic meningitis, vasculitis and encephalitis.
Besides fungal infections, CNS may be involved in protozoal diseases such as in malaria, toxoplasmosis, amoebiasis, trypanosomiasis and cysticerosis (Fig. 30.6).
CEREBROVASCULAR DISEASES
Cerebrovascular diseases are all those diseases in which one or more of the blood vessels of the brain are involved in the pathologic processes. Various pathologic processes commonly implicated in cerebrovascular diseases are: thrombosis, embolism, rupture of a vessel, hypoxia, hypertensive arteriolosclerosis, atherosclerosis, arteritis, trauma, aneurysm and developmental malformations. These processes can result in 2 main types of parenchymal diseases of the brain:
A. Ischaemic brain damage:
a)Generalised reduction in blood flow resulting in global hypoxic-ischaemic encephalopathy
b)Local vascular obstruction causing infarcts.
B. Intracranial haemorrhage: |
879 |
a)Haemorrhage in the brain parenchyma (intracerebral haemorrhage)
b)Haemorrhage in the subarachnoid space (subarachnoid
haemorrhage).
The stroke syndrome is the cardinal feature of cerebrovascular disease. The term stroke is used for sudden and dramatic development of focal neurologic deficit, varying from trivial neurologic disorder to hemiplegia and coma. Other less common effects of vascular disease include: transient ischaemic attacks (TIA), vascular headache (e.g. in migraine, hypertension and arteritis), local pressure of an aneurysm and increased intracranial pressure (e.g. in hypertensive encephalopathy and venous thrombosis).
A few important forms are discussed below.
A. ISCHAEMIC BRAIN DAMAGE |
|
|
Ischaemic necrosis in the brain results from ischaemia caused |
|
|
by considerable reduction or complete interruption of blood |
|
|
supply to neural tissue which is insufficient to meet its |
|
|
metabolic needs. The brain requires sufficient quantities of |
CHAPTER |
|
oxygen and glucose so as to sustain its aerobic metabolism, |
||
mainly by citric acid (Krebs’) cycle which requires oxygen. |
||
|
||
Moreover, neural tissue has limited stores of energy reserves |
|
|
so that cessation of continuous supply of oxygen and glucose |
|
|
for more than 3-4 minutes results in permanent damage to |
|
|
neurons and neuroglial cells. |
30 |
|
Deprivation of oxygen (anoxia) to the brain may occur in |
||
4 different ways: |
|
|
1. Anoxic anoxia, in which there is low inspired pO2. |
The |
|
2. Anaemic anoxia, in which the oxygen-carrying haemo- |
||
globin is reduced. |
Nervous |
|
4. Stagnant (ischaemia) anoxia, in which the damage is caused |
||
3. Histotoxic anoxia, in which there is direct toxic injury as |
|
|
occurs in cyanide poisoning. |
|
|
by cessation of blood with resultant local accumulation of |
System |
|
metabolites and changes in pH. |
||
|
||
In all these different forms of anoxia, the end-result is |
|
|
ischaemic brain damage which may have one of the following |
|
|
two patterns: |
|
|
1. Global hypoxic-ischaemic encephalopathy, resulting from |
|
|
generalised cerebral hypoperfusion. |
|
|
2. Cerebral infarction, resulting from severe localised |
|
|
reduction or cessation of blood supply. |
|
|
Global Hypoxic-Ischaemic Encephalopathy |
|
|
The brain receives 20% of cardiac output for maintaining its |
|
|
vital aerobic metabolism. A number of factors determine the |
|
|
maximum length of time the CNS can survive irreversible |
|
|
ischaemic damage. These are as under: |
|
i)Severity of the hypoxic episode.
ii)Presence of pre-existing cerebrovascular disease.
iii)Age of the patient.
iv)Body temperature.
In normal individuals, the brain continues to be perfused
adequately up to systolic arterial pressure of 50 mmHg by an auto-regulatory vascular control mechanism. However,

880fall of systemic arterial systolic pressure below this critical value results in rapid fall in cerebral perfusion pressure and eventual ischaemic encephalopathy. Such types of medical emergencies occur at the time of cardiac arrest followed by relatively delayed resuscitation, severe episode of hypotension, carbon monoxide intoxication and status epilepticus. Hypoxic encephalopathy may be followed by a postischaemic confusional state and complete recovery or a state of coma and even a persistent vegetative life and brain death.
Depending upon the proneness of different cells of the brain to the effects of ischaemia-hypoxia, three types of lesion may occur:
1. Selective neuronal damage: Neurons are most vulnerable to damaging effect of ischaemia-hypoxia and irreversible injury. In particular, oligodendroglial cells are most susceptible, followed by astrocytes while microglial cells and
vascular endothelium survive the longest. The reason for undue vulnerability of neurons to hypoxia can be explained by various factors:
i)Different cerebral circulatory blood flow.
ii)Presence of acidic excitatory neurotransmitters called
|
excitotoxins. |
|
SECTION |
iii) Excessive metabolic requirement of these neurons. |
|
iv) Increased sensitivity of neurons to lactic acid. |
||
2. Laminar necrosis: Global ischaemia of cerebral cortex |
||
results in uneven damage because of different cerebral |
||
vasculature which is termed laminar or pseudolaminar |
||
III |
necrosis. In this, superficial areas of cortical layers escape |
|
damage while deeper layers are necrosed. |
||
|
||
Systemic |
3. Watershed infarcts: Circulatory flow in the brain by |
|
suffers maximum damage. This results in wedge-shaped |
||
|
anterior, middle and posterior cerebral arteries has |
|
|
overlapping circulations. In ischaemia-hypoxia, perfusion of |
|
|
overlapping zones, being farthest from the blood supply, |
|
Pathology |
areas of coagulative necrosis called watershed or borderzone |
|
infarcts. Particularly vulnerable is the border zone of the |
||
|
||
|
cerebral cortex between the anterior and middle cerebral |
|
|
arteries, producing para-sagittal infarction. |
|
|
|
|
|
MORPHOLOGIC FEATURES. The pathologic appear- |
|
|
ance of the brain in hypoxic encephalopathy varies |
|
|
depending upon the duration and severity of hypoxic |
|
|
episode and the length of survival. |
|
|
Survival for a few hours: No pathologic changes are |
|
|
visible. |
|
|
Survival 12-24 hours: No macroscopic change is |
|
|
discernible but microscopic examination reveals early |
|
|
neuronal damage in the form of eosinophilic cytoplasm |
|
|
and pyknotic nuclei, so called red neurons. |
|
|
After 2-7 days: Grossly, there is focal softening. The |
|
|
area supplied by distal branches of the cerebral arteries |
|
|
suffers from the most severe ischaemic damage and may |
|
|
develop border zone or watershed infarcts in the junctional |
|
|
zones between the territories supplied by major arteries. |
|
|
Microscopically, the nerve cells die and disappear and |
|
|
are replaced by reactive fibrillary gliosis. There are minor |
|
|
variations in the distribution of neuronal damage to the |
|
|
|
cortex; the loss of pyramidal cell layer is more severe than that of granular cell layer producing laminar necrosis.
 Longer duration: Use of modern ventilators has led to maintenance of cardiorespiratory function in the presence of total brain necrosis unassociated with vital reaction.
Longer duration: Use of modern ventilators has led to maintenance of cardiorespiratory function in the presence of total brain necrosis unassociated with vital reaction.
Cerebral Infarction
Cerebral infarction is a localised area of tissue necrosis caused by local vascular occlusion—arterial or venous. Occasionally, it may be the result of non-occlusive causes such as compression on the cerebral arteries from outside and from hypoxic encephalopathy. Clinically, the signs and symptoms associated with cerebral infarction depend upon the region infarcted. In general, the focal neurologic deficit termed stroke, is present. However, significant atherosclerotic cerebrovascular disease may produce transient ischaemic attacks (TIA).
1. Arterial occlusion. Occlusion of the cerebral arteries by either thrombi or emboli is the most common cause of cerebral infarction. Thrombotic occlusion of the cerebral arteries is most frequently the result of atherosclerosis, and rarely, from arteritis of the cranial arteries. Embolic arterial occlusion is commonly derived from the heart, most often from mural thrombosis complicating myocardial infarction, from atrial fibrillation and endocarditis. The size and shape of an infarct are determined by the extent of anastomotic connections with adjacent arterial branches as under:

 Circle of Willis provides a complete collateral flow for internal carotid and vertebral arteries.
Circle of Willis provides a complete collateral flow for internal carotid and vertebral arteries.
 Middle and anterior cerebral arteries have partial anastomosis of their distal branches. Their complete occlusion may cause infarcts.
Middle and anterior cerebral arteries have partial anastomosis of their distal branches. Their complete occlusion may cause infarcts.

 Small terminal cerebral arteries, on the contrary, are endarteries and have no anastomosis. Hence, occlusion of these branches will invariably lead to an infarct.
Small terminal cerebral arteries, on the contrary, are endarteries and have no anastomosis. Hence, occlusion of these branches will invariably lead to an infarct.
2.Venous occlusion. Venous infarction in the brain is an infrequent phenomenon due to good communications of the cerebral venous drainage. However in cancer, due to increased predisposition to thrombosis, superior sagittal thrombosis may occur leading to bilateral, parasagittal, multiple haemorrhagic infarcts.
3.Non-occlusive causes. Compression of the cerebral arteries from outside such as occurs during herniation may cause cerebral infarction. Mechanism of watershed (border zone) cerebral infarction in hypoxic encephalopathy has already been explained above.
In any case, the extent of damage produced by any of the above causes depends upon:
i)rate of reduction of blood flow;
ii)type of blood vessel involved; and
iii)extent of collateral circulation.
MORPHOLOGIC FEATURES. Grossly, cerebral infarcts may be anaemic or haemorrhagic. An anaemic infarct
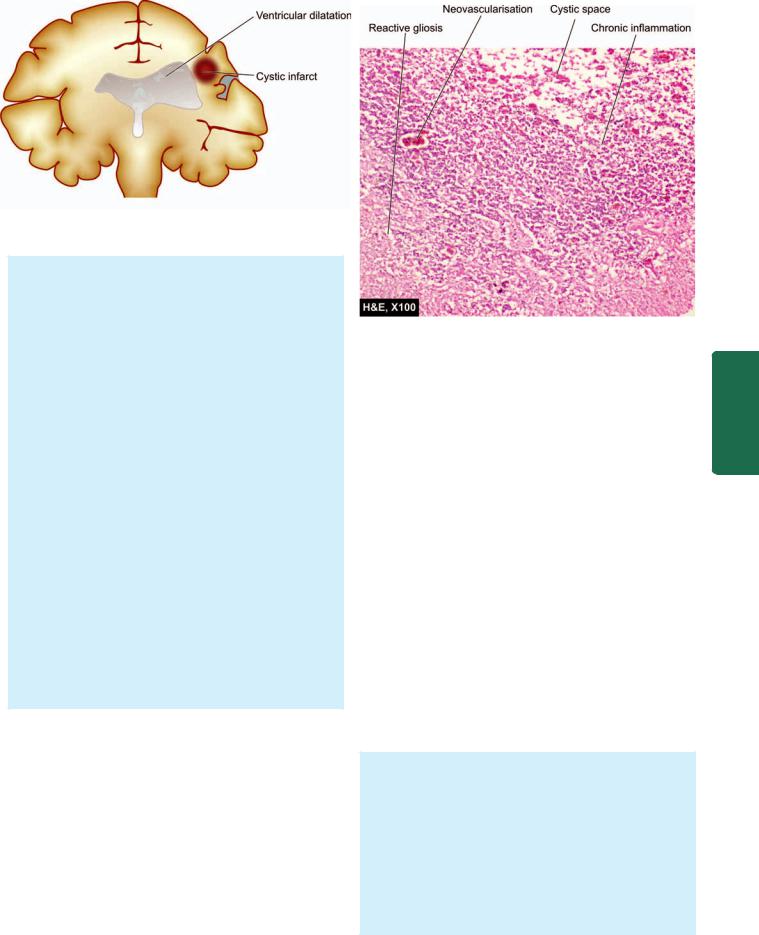
881
Figure 30.7 
 An old cystic infarct in the brain (coronal section). There is shrinkage of scarred area with ipsilateral ventricular dilatation.
An old cystic infarct in the brain (coronal section). There is shrinkage of scarred area with ipsilateral ventricular dilatation.
becomes evident 6-12 hours after its occurrence. The affected area is soft and swollen and there is blurring of junction between grey and white matter. Within 2-3 days, the infarct undergoes softening and disintegration. Eventually, there is central liquefaction with peripheral firm glial reaction and thickened leptomeninges, forming a cystic infarct (Fig. 30.7). A haemorrhagic infarct is red and superficially resembles a haematoma. It is usually the result of fragmentation of occlusive arterial emboli or venous thrombosis.
Histologically, the sequential changes are as under:
1.Initially, there is eosinophilic neuronal necrosis and lipid vacuolisation produced by breakdown of myelin. Simultaneously, the infarcted area is infiltrated by neutrophils.
2.After the first 2-3 days, there is progressive invasion by macrophages and there is astrocytic and vascular proliferation.
3.In the following weeks to months, the macrophages clear away the necrotic debris by phagocytosis followed by reactive astrocytosis, often with little fine fibrosis (Fig. 30.8). A haemorrhagic infarct has some phagocytes containing haemosiderin.
4.Ultimately, after 3-4 months an old cystic infarct is formed which shows a cyst traversed by small blood vessels and has peripheral fibrillary gliosis. Small cavitary infarcts are called lacunar infarcts and are commonly found as a complication of systemic hypertension.
B.INTRACRANIAL HAEMORRHAGE
Haemorrhage into the brain may be traumatic, non-trau- matic, or spontaneous. There are two main types of spontaneous intracranial haemorrhage:
1.Intracerebral haemorrhage, which is usually of hypertensive origin.
2.Subarachnoid haemorrhage, which is commonly aneurysmal in origin.
In addition to hypertension and rupture of an aneurysm, other causes of spontaneous intracranial haemorrhage include vascular malformations which produce mixed intracerebral and subarachnoid haemorrhage, haemorrhagic diathesis and haemorrhage into tumours.
Figure 30.8 
 An anaemic infarct of a few days duration. The histologic changes are reactive astrocytosis, a few reactive macrophages and neovascularisation in the wall of the cystic lesion. The outer cortical layer is, however, intact.
An anaemic infarct of a few days duration. The histologic changes are reactive astrocytosis, a few reactive macrophages and neovascularisation in the wall of the cystic lesion. The outer cortical layer is, however, intact.
Intracerebral Haemorrhage
Spontaneous intracerebral haemorrhage occurs mostly in patients of hypertension. Most hypertensives over middle age have microaneurysms in very small cerebral arteries in the brain tissue. Rupture of one of the numerous microaneurysms is believed to be the cause of intracerebral haemorrhage. Unlike subarachnoid haemorrhage, it is not common to have recurrent intracerebral haemorrhages.
The common sites of hypertensive intracerebral haemorrhage are the region of the basal ganglia (particularly the putamen and the internal capsule), pons and the cerebellar cortex. Clinically the onset is usually sudden with headache and loss of consciousness. Depending upon the location of the lesion, hemispheric, brainstem or cerebellar signs will be present. About 40% of patients die during the first 3-4 days of haemorrhage, mostly from haemorrhage into the ventricles. The survivors tend to have haematoma that separates the tissue planes which is followed by resolution and development of an apoplectic cyst accompanied by loss of function.
MORPHOLOGIC FEATURES. Grossly and microscopically, the haemorrhage consists of dark mass of clotted blood replacing brain parenchyma. The borders of the lesion are sharply-defined and have a narrow rim of partially necrotic parenchyma. Small ring haemorrhages in the Virchow-Robin space in the border zone are commonly present. Ipsilateral ventricles are distorted and compressed and may contain blood in their lumina. Rarely, blood may rupture through the surface of the brain into the subarachnoid space. After a few weeks to months, the haematoma undergoes resolution with formation of a
System Nervous The 30 CHAPTER
882slit-like space called apoplectic cyst which contains yellowish fluid. Its margins are yellow-brown and have haemosiderin-laden macrophages and a reactive zone of fibrillary astrocytosis.
|
Subarachnoid Haemorrhage |
|
|
Haemorrhage into the subarachnoid space is most comm- |
|
|
only caused by rupture of an aneurysm, and rarely, rupture |
|
|
of a vascular malformation. |
|
|
A general discussion of aneurysms is given on page 405. |
|
|
Of the three types of aneurysms affecting the larger |
|
|
intracranial arteries—berry, mycotic and fusiform, berry |
|
|
aneurysms are most important and most common. |
|
|
BERRY ANEURYSMS are saccular in appearance with |
|
|
rounded or lobulated bulge arising at the bifurcation of |
|
|
intracranial arteries and varying in size from 2 mm to 2 cm |
|
|
or more. They account for 95% of aneurysms which are liable |
|
|
to rupture. Berry aneurysms are rare in childhood but |
|
|
increase in frequency in young adults and middle life. They |
|
|
are, therefore, not congenital anomalies but develop over the |
|
|
years from developmental defect of the media of the arterial |
|
SECTION |
wall at the bifurcation of arteries forming thin-walled saccu- |
|
lar bulges. Although most berry aneurysms are sporadic in |
||
|
||
|
occurrence, there is an increased incidence of their presence |
|
|
in association with congenital polycystic kidney disease and |
|
|
coarctation of the aorta. About a quarter of berry aneurysms |
|
III |
are multiple. |
|
In more than 85% cases of subarachnoid haemorrhage, |
||
|
the cause is massive and sudden bleeding from a berry |
|
Systemic |
aneurysm on or near the circle of Willis. The four most |
|
the stem of the internal carotid artery. |
||
|
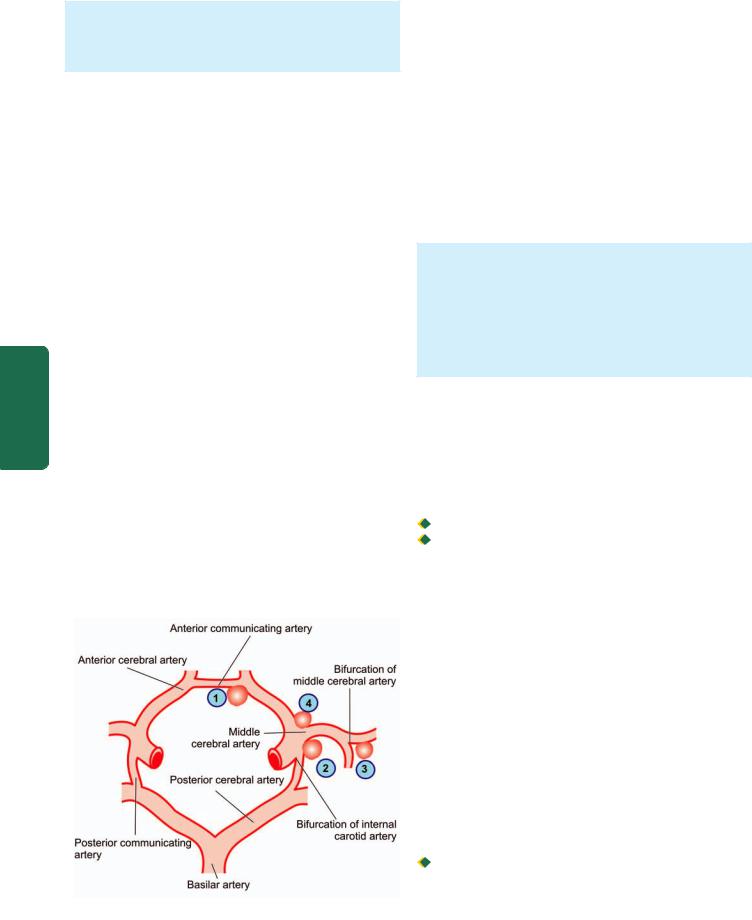
common sites are as under (Fig. 30.9): |
|
|
1. In relation to anterior communicating artery. |
|
|
2. At the origin of the posterior communicating artery from |
|
Pathology |
3. At the first major bifurcation of the middle cerebral artery. |
|
4. At the bifurcation of the internal carotid into the middle |
||
|
||
|
and anterior cerebral arteries. |
Figure 30.9 
 The circle of Willis showing principal sites of berry
The circle of Willis showing principal sites of berry
(saccular) aneurysms. The serial numbers indicate the frequency of involvement.
The remaining 15% cases of subarachnoid haemorrhage are the result of rupture in the posterior circulation, vascular malformations and rupture of mycotic aneurysms that occurs in the setting of bacterial endocarditis. In all types of aneurysms, the rupture of thin-walled dilatation occurs in association with sudden rise in intravascular pressure but chronic hypertension does not appear to be a risk factor in their development or rupture.
Clinically, berry aneurysms remain asymptomatic prior to rupture. On rupture, they produce severe generalised headache of sudden onset which is frequently followed by unconsciousness and neurologic defects. Initial mortality from first rupture is about 20-25%. Survivors recover completely but frequently suffer from recurrent episodes of fresh bleeding.
MORPHOLOGIC FEATURES. Rupture of a berry aneurysm frequently spreads haemorrhage throughout the subarachnoid space with rise in intracranial pressure and characteristic blood-stained CSF. An intracerebral haematoma may develop if the blood tracks into the brain parenchyma. The region of the brain supplied by the affected artery frequently shows infarction, partly attributed to vasospasm.
TRAUMA TO THE CNS
Trauma to the CNS constitutes an important cause of death and permanent disability in the modern world. Important causes of head injuries are: motor vehicle accidents, accidental falls and violence. Traumatic injuries to the CNS may result in three consequences which may occur in isolation or in combination:

 epidural haematoma;
epidural haematoma;

 subdural haematoma; and
subdural haematoma; and 
 parenchymal brain damage.
parenchymal brain damage.
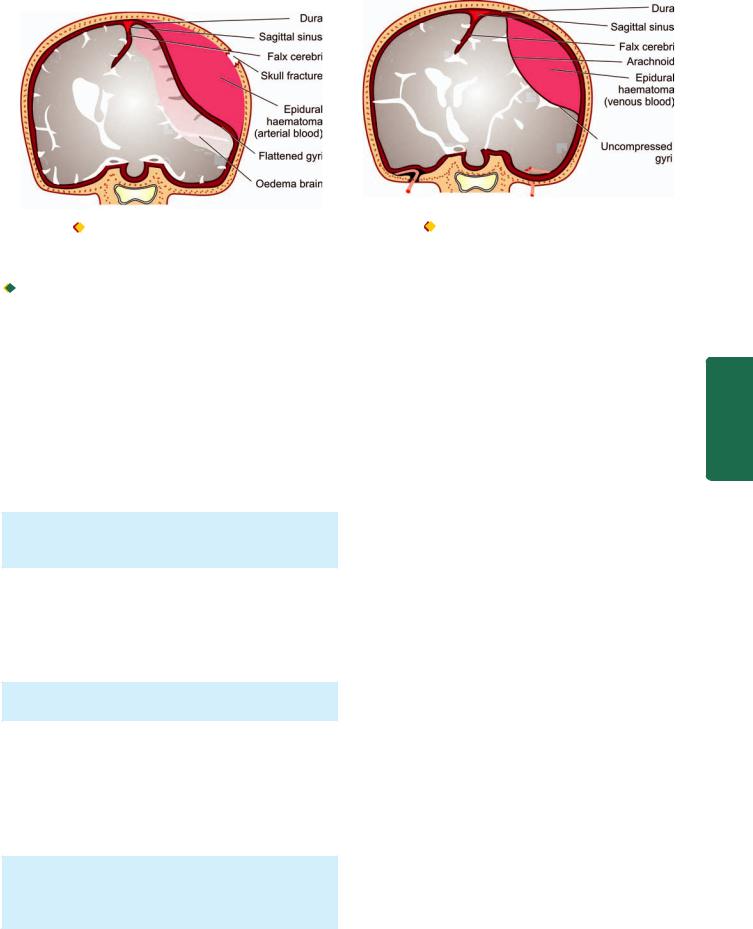
A. Epidural Haematoma
Epidural haematoma is accumulation of blood between the dura and the skull following fracture of the skull, most commonly from rupture of middle meningeal artery. The haematoma expands rapidly since accumulating blood is arterial in origin and causes compression of the dura and flattening of underlying gyri (Fig. 30.10). The patient develops progressive loss of consciousness if haematoma is not drained early.
B. Subdural Haematoma
Subdural haematoma is accumulation of blood between the dura and subarachnoid and develops most often from rupture of veins which cross the surface convexities of the cerebral hemispheres. Subdural haematoma may be acute or chronic.

 Acute subdural haematoma. Acute subdural haematoma develops following trauma and consists of clotted blood, often in the frontoparietal region. There is no significant compression of gyri (Fig. 30.11). Since the accumulated blood is of venous origin, symptoms appear slowly and may become chronic with passage of time if not fatal.
Acute subdural haematoma. Acute subdural haematoma develops following trauma and consists of clotted blood, often in the frontoparietal region. There is no significant compression of gyri (Fig. 30.11). Since the accumulated blood is of venous origin, symptoms appear slowly and may become chronic with passage of time if not fatal.
883
Figure 30.10 |
Epidural haematoma often results from rupture of |
Figure 30.11 |
Subdural haematoma often results from rupture of |
artery following skull fracture resulting in accumulation of arterial blood |
veins crossing the cerebral convexities and is characterised by |
||
between the skull and the dura. |
accumulation of venous blood between the dura and the arachnoid. |
||

 Chronic subdural haematoma. Chronic subdural haematoma occurs often with brain atrophy and less commonly following trauma. Chronic subdural haematoma is composed of liquid blood. Separating the haematoma from underlying brain is a membrane composed of granulation tissue.
Chronic subdural haematoma. Chronic subdural haematoma occurs often with brain atrophy and less commonly following trauma. Chronic subdural haematoma is composed of liquid blood. Separating the haematoma from underlying brain is a membrane composed of granulation tissue.
C. Parenchymal Brain Damage
Trauma to the CNS may result in damage to brain parenchyma and includes the following forms:
1. Concussion. Concussion is caused by closed head injury and is characterised by transient neurologic dysfunction and loss of consciousness. Invariably, there is complete neurologic recovery after some hours to days.
No significant morphologic change is noticed but more severe concussion may cause diffuse axonal injury (discussed below).
2. Diffuse axonal injury. Diffuse axonal injury is the most common cause of persistent coma or vegetative state following head injury. The underlying cause is sudden angular acceleration or deceleration resulting in widespread axonal shearing in the deep white matter of both the hemispheres.
Grossly, the changes are minimal to small multiple haemorrhages.
3. Contusions and lacerations. Contusions and lacerations are the result of direct damage to the brain parenchyma, particularly cerebral hemispheres, as occurs in the soft tissues. Most often, they are the result of blunt trauma. The overlying skull may or may not be fractured. Traumatic subarachnoid haemorrhage invariably accompanies cerebral contusions.
Microscopically, brain parenchyma at the affected site is haemorrhagic, necrotic and fragmented. On healing, these lesions appear as shrunken areas with golden brown haemosiderin pigment on the surface.
4.Traumatic intracerebral haemorrhage. On trauma to the CNS, the parenchymal vessels of the hemispheres may get torn and cause multiple intracerebral haemorrhages.
5.Brain swelling. Head injury may be accompanied by localised or diffuse brain swelling.
DEMYELINATING DISEASES
Demyelinating diseases are an important group of neurological disorders which have, in common, the pathologic features of focal or patchy destruction of myelin sheaths in the CNS accompanied by an inflammatory response. Demyelination may affect peripheral nervous system as well. Some degree of axonal damage may also occur but demyelination is the predominant feature. The exact cause for demyelination is not known but currently viral infection and autoimmunity are implicated in its pathogenesis.
Loss of myelin may occur in certain other conditions as well, but without an inflammatory response. These conditions have known etiologies such as: geneticallydetermined defects in the myelin metabolism (leucodystrophies), slow virus diseases of oligodendrocytes (progressive multifocal leucoencephalopathy), and exposure to toxins (central pontine myelinolysis). All these entities are currently not classified as demyelinating diseases. Only those conditions in which the myelin sheath or the myelin-forming cells (i.e. oligodendrocytes and Schwann cells) are primarily injured and are associated with considerable inflammatory exudate are included under the term ‘demyelinating diseases’. Pathologically and clinically, two demyelinating diseases are distinguished:
1.Multiple or disseminated sclerosis
2.Perivenous encephalomyelitis.
Multiple (Disseminated) Sclerosis
Multiple or disseminated sclerosis is the most common of the CNS demyelinating diseases. The usual age at onset is 20 to 40 years. The disease presents as recurrent attacks of focal neurologic disorder with predilection for involvement
System Nervous The 30 CHAPTER

884of the spinal cord, optic nerve and brain. The first attack usually begins with a single sign or symptom, most commonly optic neuritis, followed by recovery. As the disease becomes more progressive, remissions become infrequent and incomplete. The etiology of multiple sclerosis remains unknown but a role for genetic susceptibility, infectious agent and immunologic mechanism has been proposed.
|
MORPHOLOGIC FEATURES. The pathologic hallmark |
|
|
is the presence of many scattered discrete areas of |
|
|
demyelination termed plaques. |
|
|
Grossly, plaques appear as grey-pink, swollen, sharply |
|
|
defined, usually bilaterally symmetric areas in the white |
|
|
matter. |
|
|
Microscopically, the features vary according to the age |
|
|
of the plaque: |
|
|
1. ln active enlarging plaques, the histologic features are |
|
|
accumulation of lymphocytes and macrophages around |
|
|
venules and at the plaque margin where demyelination |
|
|
is occurring. In addition, there is loss of oligodendrocytes |
|
SECTION |
and presence of reactive astrocytosis with numerous lipid- |
|
laden macrophages (microglia) in the plaque. The axons |
||
|
||
|
in the plaque are generally intact. |
|
|
2. In old inactive plaques, there is no perivascular inflam- |
|
|
matory cell infiltrate and nearly total absence of |
|
III |
oligodendrocytes. Demyelination in the plaque area is |
|
complete as there is only limited regeneration of myelin. |
||
|
Gliosis is well-developed but astrocytes are less |
|
Systemic |
prominent. Some axonal loss may be present. |
|
Perivenous Encephalomyelitis |
||
|
||
|
Perivenous encephalomyelitis includes two uncommon |
|
Pathology |
diseases: acute disseminated encephalomyelitis and acute |
|
necrotising haemorrhagic leucoencephalitis. Both are monophasic |
||
|
||
|
diseases characterised by perivenous mononuclear |
|
|
inflammatory cell infiltration. Both diseases occur following |
|
|
a viral infection, vaccination or a respiratory illness. Both |
|
|
these conditions are looked upon as human counterpart of |
|
|
experimental allergic encephalomyelitis (EAE) and are |
|
|
considered to be allergic reaction against myelin antigen. |
|
|
Acute disseminated encephalomyelitis occurs usually |
|
|
following viral infection (measles, mumps, rubella, |
|
|
chickenpox), whooping cough or vaccination. The disease |
|
|
begins abruptly with headache and delirium followed by |
|
|
lethargy and coma. Signs of meningeal irritation and fever |
|
|
may be present. Prognosis for recovery is generally good. |
|
|
Acute necrotising haemorrhagic leucoencephalitis is a |
|
|
rare disease occurring more often after a respiratory infection. |
|
|
The clinical course is similar to that of acute disseminated |
|
|
encephalomyelitis except for its suddenness of onset and |
|
|
rapidity of progression, sometimes leading to death within |
|
|
48 hours. |
|
|
MISCELLANEOUS DISEASES |
|
|
Included under the heading of miscellaneous diseases of the |
|
|
CNS are degenerative, metabolic and nutritional diseases. |
These groups along with the list of diseases included in each group are briefly outlined below without going into the details of individual diseases for which the interested reader may consult pertinent text on neuropathology and neurology.
Degenerative Diseases
Degenerative diseases are disorders of unknown etiology and pathogenesis, characterised pathologically by progressive loss of CNS neurons and their processes accompanied by fibrillary astrocytosis. The identification of these diseases depends upon exclusion of diseases with known etiologies such as metabolic disturbances, vascular diseases, nutritional deficiencies or infection. A considerable proportion of degenerative disorders are genetic in origin, with either dominant or recessive inheritance; others occur sporadically. Family history is, of course, of great importance.
The degenerative disorders usually begin insidiously and have a gradual progressive course over many years. In virtually all cases, the lesions have characteristic bilaterally symmetric distribution. Another striking characteristic of the degenerative disorders is that particular anatomic or physiologic system of neurons may be selectively affected, leaving others entirely intact.
Classification of degenerative diseases into individual syndromes is based on clinical aspects and anatomic distribution of the lesions. Some of the more common degenerative diseases are listed in Table 30.3. Two of the important examples—Alzheimer’s disease and parkinsonism, are considered below.
ALZHEIMER’S DISEASE. Alzheimer’s disease is the most common cause of dementia in the elderly. The condition occurs after 5th decade of life and its incidence progressively increases with advancing age. The exact cause is not known but a few factors are implicated in its etiology which include positive family history and deposition of Aβ amyloid derived from amyloid precursor protein (APP) forming neuritic ‘senile’
plaques and neurofibrillary tangles.
Grossly, the brain is often reduced in weight and bilaterally atrophic.
Microscopically, the main features are as under:
i)Senile neuritic plaque is the most conspicuous lesion and consists of focal area which has a central core containing Aβ amyloid.
ii)Neurofibrillary tangle is a filamentous collection of neurofilaments and neurotubules within the cytoplasm of neurons.
iii)Amyloid angiopathy is deposition of the same amyloid in the vessel wall which is deposited in the amyloid core of the plaque.
iv)Granulovacuolar degeneration is presence of multiple, small intraneuronal cytoplasmic vacuoles, some of which contain one or more dark granules called Hirano bodies.
PARKINSONISM. Parkinsonism is a syndrome of chronic progressive disorder of motor function and is clinically characterised by tremors which are most conspicuous at rest and worsen with emotional stress; other features are rigidity
TABLE 30.3: Common Degenerative Diseases.
Region Affected |
Disease |
Main Features |
Predominant Pathology |
|
|
|
|
I. Cerebral cortex |
Alzheimer’s disease |
Progressive senile |
Cortical atrophy, senile plaques (neurites), |
|
|
dementia |
neurofibrillary tangles, amyloid angiopathy |
|
Pick’s disease |
Pre-senile dementia |
Lobar cortical atrophy, ballooning degeneration |
|
|
|
of neurons (Pick’s cells) |
II. Basal ganglia and |
Huntington’s disease |
Progressive dementia with |
Atrophy of frontal lobes, fibrillary astrocytosis |
brainstem |
|
choreiform movements |
|
|
Parkinson’s disease |
Abnormalities of posture |
Aggregates of melanin-containing nerve cells in |
|
|
and movement |
brainstem, intracytoplasmic neuronal inclusions |
|
|
|
(Lewy bodies) |
III. Spinal cord and |
Cerebellar cortical |
Progressive cerebellar |
Loss of Purkinje cells in cerebellar cortex |
cerebellum |
degeneration |
ataxia |
|
|
Olivopontocerebellar |
Cerebellar ataxia |
Combination of atrophy of cerebellar cortex, |
|
atrophy |
|
inferior olivary nuclei and pontine nuclei |
|
Spinocerebellar atrophy |
Gait ataxia, dysarthria |
Degeneration of spinocerebellar tracts, |
|
(Friedreich’s ataxia) |
|
peripheral axons and myelin sheaths |
IV. Motor neurons |
Motor neuron disease |
Syndromes of muscular |
Progressive loss of motor neurons, both in the |
(UMN and LMN) |
(Amyotrophic lateral |
weakness and wasting |
cerebellar cortex (UMN) and in the anterior |
|
sclerosis) |
without sensory loss |
horn of spinal cord (LMN) |
|
Werdnig-Hoffmann’s |
Spinal muscular |
Loss of lower motor neurons, |
|
disease |
atrophy in infants |
denervation atrophy of muscles |
|
|
|
|
and disordered gait and posture. Parkinsonism is caused by several degenerative diseases, the most important being Parkinson’s disease; other causes of parkinsonism are trauma, toxic agents, and drugs (dopamine antagonists).
Grossly, the brain is atrophic or may be normal externally. Microscopically, the hallmark is depigmentation of substantia nigra and locus ceruleus due to loss of neuromelanin pigment from neurons and accumulation of neuromelanin pigment in the glial cells. Some of the residual neurons in these areas contain intracytoplasmic, eosinophilic, elongated inclusions called Lewy bodies.
Metabolic Diseases
Metabolic diseases of the CNS result from neurochemical disturbances which are either inherited or acquired.
Hereditary metabolic disorders predominantly manifest in infancy or childhood and include genetically-determined disorders of carbohydrate, lipid, amino acid and mineral metabolism. Acquired or secondary metabolic diseases are the disturbances of cerebral function due to disease in some other organ system such as the heart and circulation, lungs and respiratory function, kidneys, liver, endocrine glands and pancreas. In addition, endogenous metabolic diseases may be caused by toxic injuries induced by metals, gases, chemicals, and drugs. The pathologic changes in each of these conditions are quite diverse and include oedema, neuronal storage, degenerative changes, and sometimes parenchymal necrosis.
The predominant types of hereditary and acquired metabolic disorders are as under:
A. HEREDITARY METABOLIC DISEASES:
1.Neuronal storage diseases—characterised by storage of a metabolic product in the neurons due to specific enzyme deficiency. Common examples are: gangliosidoses (e.g. Tay-Sachs disease or GM2 gangliosidosis), mucopolysaccharidoses, Gaucher’s disease and Niemann-Pick disease). These conditions are described on page 262.
2.Leucodystrophies—are diseases of white matter characterised by diffuse demyelination and gliosis. They are caused by deficiency of one of the enzymes required for formation and maintenance of myelin. That is why these conditions are also called dysmyelinating diseases. Common types of leucodystrophies are: sudanophilic leucodystrophy, adrenoleucodystrophy, metachromatic leucodystrophy and globoid cell leucodystrophy (Krabbe’s disease).
3.Other inborn errors of metabolism—e.g. Wilson’s disease (hepatolenticular degeneration), glycogen-storage diseases, phenylketonuria and galactosaemia.
B. ACQUIRED METABOLIC DISEASES:
These include the following:
1.Anoxic-ischaemic encephalopathy
2.Hypoglycaemic encephalopathy
3.Hyperglycaemic coma
4.Acute hepatic encephalopathy (Reye’s syndrome)
5.Chronic hepatic encephalopathy
6.Kernicterus
7.Uraemic encephalopathy
8.Encephalopathy due to electrolyte and endocrine disturbances.
All these conditions have already been discussed in the relevant chapters.
885
System Nervous The 30 CHAPTER

886Nutritional Diseases
Neurologic disorders may be caused by malnutrition from lack of adequate diet in many developing countries and many poor socio-economic groups. In the United States and Europe, however, nutritionally-induced disease is chiefly found in association with chronic alcoholism or due to defect in absorption, transport or metabolism of dietary nutrients.
The general aspects of deficiency diseases have been covered in Chapter 9. Some of the common neurologic diseases included in the category of deficiency diseases are as under:
1.Wernicke’s encephalopathy and Korsakoff’s psychosis (vitamin B1 or thiamine deficiency).
2.Subacute combined degeneration of the spinal cord Figure 30.12  The anatomic distribution of common intracranial
The anatomic distribution of common intracranial
Pathology Systemic III SECTION
12deficiency).
3.Folic acid deficiency (page 304).
4.Spinocerebellar syndrome (vitamin E deficiency).
5.Pellagra (niacin deficiency).
6.Alcoholic cerebellar degeneration.(vitamin B
TUMOURS OF THE CNS
Tumours of the CNS may originate in the brain or spinal cord (primary tumours), or may spread to the brain from another primary site of cancer (metastatic tumours). More than one-quarter of the CNS tumours are secondary metastases arising in patients undergoing treatment for systemic cancer. Primary CNS tumours are the second commonest form of cancer in infants and children under the age of 15 years, exceeded in frequency only by leukaemia. Both benign and malignant CNS tumours are capable of producing neurologic impairment depending upon their site.
Primary CNS tumours or intracranial tumours include: tumours arising from constituent cells of the brain (with the sole exception of microglial cells) and from the supporting tissues. Childhood brain tumours arise from more primitive cells (e.g. neuroblastoma, medulloblastoma).
A classification of intracranial tumours abbreviated from the WHO classification is given in Table 30.4. The anatomic distribution of common intracranial tumours is illustrated in Fig. 30.12. Among the primary brain tumours, gliomas constitute 50-60%, meningiomas 25%, schwannomas 10% and other primary tumours comprise the remainder.
Some of the important morphologic types are described below.
tumours.
Gliomas may be well-differentiated or poorly-differen- tiated. However, gliomas are never truly well-demarcated or encapsulated and thus all grades of gliomas infiltrate the adjacent brain tissue. Gliomas are disseminated to other parts of the CNS by CSF but they rarely ever metastasise beyond the CNS.
TABLE 30.4: Classification of Intracranial Tumours.
I. TUMOURS OF NEUROGLIA (GLIOMAS)
1.Astrocytoma
2.Oligodendroglioma
3.Ependymoma
4.Choroid plexus papilloma
II.TUMOURS OF NEURONS
1.Neuroblastoma (page 800)
2.Ganglioneuroblastoma
3.Ganglioneuroma
III.TUMOURS OF NEURONS AND NEUROGLIA Ganglioglioma
IV. POORLY-DIFFERENTIATED AND EMBRYONAL TUMOURS
1.Medulloblastoma
2.Neuroblastoma (page 800)
3.PNET (page 848)
V.TUMOURS OF MENINGES
1.Meningioma
2.Meningeal sarcoma
VI. NERVE SHEATH TUMOURS
1.Schwannoma (neurilemmoma)
2.Neurofibroma
3.Malignant nerve sheath tumour
GLIOMAS
The term glioma is used for all tumours arising from neuroglia, or more precisely, from neuroectodermal epithelial tissue. Gliomas are the most common of the primary CNS tumours and collectively account for 40% of all intracranial tumours. They include tumours arising:
 from astrocytes (astrocytomas and glioblastoma multiforme);
from astrocytes (astrocytomas and glioblastoma multiforme);

 from oligodendrocytes (oligodendroglioma);
from oligodendrocytes (oligodendroglioma);

 from ependyma (ependymoma); and
from ependyma (ependymoma); and

 from choroid plexus (choroid plexus papilloma).
from choroid plexus (choroid plexus papilloma).
VII. OTHER PRIMARY INTRAPARENCHYMAL TUMOURS
1.Haemangioblastoma
2.Primary CNS lymphoma
3.Germ cell tumours
VIII. MISCELLANEOUS TUMOURS
1.Malignant melanoma (page 787)
2.Craniopharyngioma (page 796)
3.Pineal cell tumours
4.Pituitary tumours
IX. TUMOUR-LIKE LESIONS
(epidermal cyst, dermoid cyst, colloid cyst)
X.METASTATIC TUMOURS
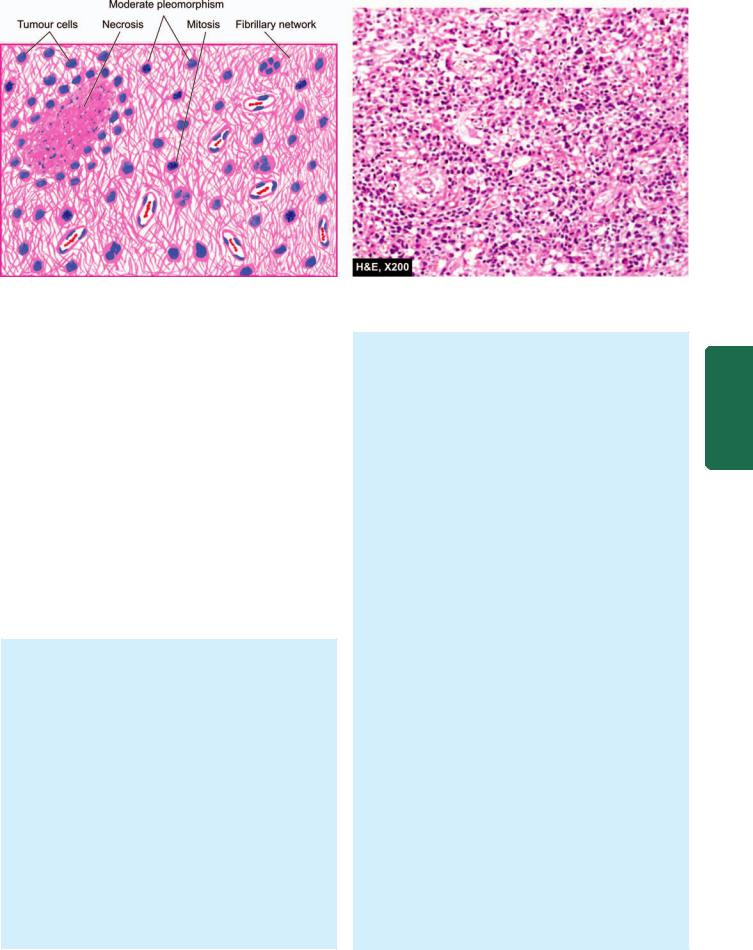
887
Figure 30.13 
 Anaplastic astrocytoma, showing hypercellularity of pleomorphic astrocytic cells, mitoses and vascular proliferation in fibrillary background. Areas of necrosis are also present.
Anaplastic astrocytoma, showing hypercellularity of pleomorphic astrocytic cells, mitoses and vascular proliferation in fibrillary background. Areas of necrosis are also present.
Astrocytomas (including Glioblastoma Multiforme)
Astrocytomas are the most common type of gliomas. In general, they are found in the late middle life with a peak in 6th decade of life. They occur predominantly in the cerebral hemispheres, and occasionally in the spinal cord. In children and young adults, pilocytic astrocytomas arise in the optic nerves, cerebellum and brainstem. Astrocytomas have tendency to progress from low grade to higher grades of anaplasia. Low-grade astrocytomas evolve slowly over several years whereas higher grades (anaplastic astrocytoma and glioblastoma multiforme) bring about rapid clinical deterioration of the patient.
The diagnosis of various types of astrocytomas can be generally made by routine H & E morphology but in difficult situations and poorly differentiated cases, immunohistochemical staining with glial fibrillary protein (GFAP) or by electron microscopic demonstration of glial filaments can be done.
MORPHOLOGIC FEATURES. Pathologically, astrocytomas have been conventionally divided into 3 progressive histologic grades: fibrillary (most common), gemistocytic and protoplasmic. However, currently WHO classification of astrocytomas is widely used which divides them into 4 grades from grade I (low grade) to grade IV (glioblastoma multiforme) as under.
WHO GRADE I ASTROCYTOMA. Also called as diffuse astrocytoma, it is a low-grade tumour having good prognosis and includes special histologic entities which mainly occur in children as under:
i) Juvenile pilocytic astrocytoma. It occur in children and young adults in the cerebellum, third ventricle and optic nerve pathway.
Grossly, it is usually cystic or solid and circumscribed. Microscopically, it is predominantly composed of fusiform pilocytic astrocytes having unusually long, wavy fibrillary processes.
ii) Pleomorphic xanthoastrocytoma. It looks histologically pleomrphic and alarming but has favourable prognosis.
WHO GRADE II (WELL-DIFFERENTIATED) ASTROCYTOMA. It is also called as fibrillary astrocytoma and is the most common form of glioma occurring in 3rd to 4th decades of life.
Grossly, it is a poorly defined, grey-white tumour of variable size. The tumour distorts the underlying brain tissue and merges with the surrounding tissue.
Histologically, it is composed of well-differentiated astrocytes separated by variable amount of fibrillary background of astrocytic processes. Based on the type of astrocytes, three subtypes are distinguished: fibrillary,
protoplastic and gemistocytic astrocytoma.
WHO GRADE III (ANAPLASTIC) ASTROCYTOMA.
It generally evolves from lower grade of astrocytoma.
Grossly, it may not be distinguishable from the low-grade astrocytoma.
Histologically, it contains features of anaplasia such as hypercellularity, pleomorphism, nuclear hyperchromatism and mitoses. Another characteristic feature of anaplastic variety of astrocytoma is the proliferation of vascular endothelium (Fig. 30.13).
WHO GRADE IV ASTROCYTOMA (GLIOBLASTOMA MULTIFORME). Although its nomenclature means its origin from embryonal cells but now it is known that this tumour arises by neoplastic transformation of mature astrocytes. It is the most aggressive of astrocytomas.
Grossly, it shows variegated appearance, with some areas showing grey-white appearance while others are yellow and soft with foci of haemorrhages and necrosis. The surrounding normal brain tissue is distorted and infiltrated by yellow tumour tissue.
System Nervous The 30 CHAPTER
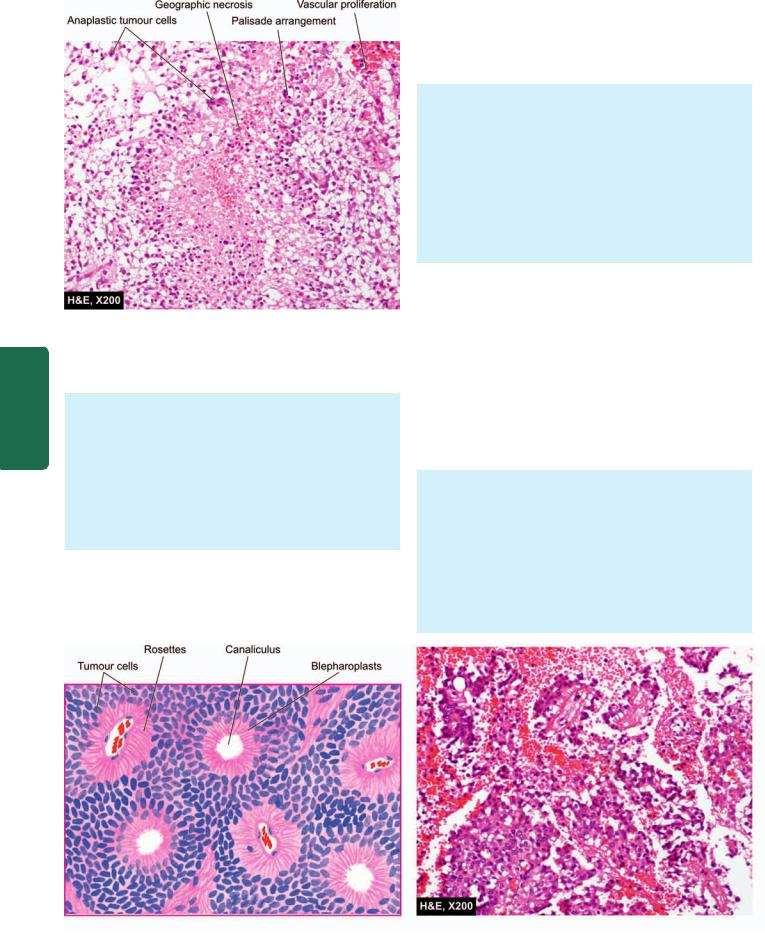
888
Pathology Systemic III SECTION
Figure 30.14 
 Glioblastoma multiforme (WHO grade IV astrocytoma). The tumour is densely cellular having marked pleomorphism.
Glioblastoma multiforme (WHO grade IV astrocytoma). The tumour is densely cellular having marked pleomorphism.
Characteristically, the tumour has areas of necrosis which are surrounded by a palisade layer of tumour cells.
Histologically, the features are as under (Fig. 30.14):
i)It has highly anaplastic and cellular appearance. The cell types show marked variation consisting of fusiform cells, small poorly-differentiated round cells, pleomorphic cells and giant cells. Mitoses are quite frequent and glial fibrils are scanty.
ii)It shows areas of tumour necrosis around which tumour cells may form pseudopalisading.
iii)Microvascular endothelial proliferation is marked.
Oligodendroglioma
Oligodendroglioma is an uncommon glioma of oligodendroglial origin and may develop in isolation or may be mixed with other glial cells. The tumour commonly presents
in 3rd to 4th decades of life. It occurs in the cerebral hemispheres, most commonly in the frontal lobes or within the ventricles. X-ray examination and CT scan reveal a welldefined mass with numerous small foci of calcification. The tumour is generally slow-growing.
MORPHOLOGIC FEATURES. Grossly, oligodendroglioma is well-circumscribed, grey-white and gelatinous mass having cystic areas, foci of haemorrhages and calcification.
Microscopically, the tumour is characterised by uniform cells with round to oval nuclei surrounded by a clear halo of cytoplasm and well-defined cell membranes. Tumour cells tend to cluster around the native neurons forming satellitosis. Typically, there are varying degree of endothelial cell hyperplasia and foci of calcification. Anaplastic change may occur as in other gliomas.
Ependymoma
Ependymoma is not an uncommon tumour, derived from the layer of epithelium that lines the ventricles and the central canal of the spinal cord. It occurs chiefly in children and young adults (below 20 years of age). Typically, it is encountered in the fourth ventricle (posterior fossa tumour). Other locations are the lateral ventricles, the third ventricle, and in the case of adults, the spinal cord in the region of lumbar spine. Clinically, by virtue of their frequent location in the floor of the fourth ventricle, ependymomas are associated with obstructive hydrocephalus. The usual biologic behaviour is of a slow-growing tumour over a period of years.
MORPHOLOGIC FEATURES. Grossly, ependymoma is a well-demarcated tumour but complete surgical removal may not be possible due to close proximity to vital structures in the medulla and pons.
Microscopically, the tumour is composed of uniform epithelial (ependymal) cells forming rosettes, canals and perivascular pseudorosettes. By light microscopy under high magnification, PTAH-positive structures, blepharoplasts, representing basal bodies of cilia may be demonstrated in the cytoplasm of tumour cells (Fig. 30.15).
Figure 30.15 
 Ependymoma showing uniform ependymal tumour cells forming rosettes and canaliculi.
Ependymoma showing uniform ependymal tumour cells forming rosettes and canaliculi.

Most tumours are well-differentiated but anaplastic variants are also recognised.
Two variants of ependymoma deserve special mention: myxopapillary type and subependymoma.
Myxopapillary ependymoma. It is a special variant of ependymoma which is common and occurs in adults. Characteristically, it occurs in the region of cauda equina and originates from the filum terminale. True to its name, it contains myxoid and papillary structures interspersed in the typical ependymal cells. It is a slow-growing tumour having a better prognosis.
Subependymoma. It occurs as a small, asymptomatic, incidental solid nodule in the fourth and lateral ventricle of middle-aged or elderly patients. Areas of microcysts and calcification may be encountered.
Histologically, it is composed of nests of uniform ependymal cells in a stroma of very dense, acellular, finely fibrillar background. Subependymoma is typically a very slow-growing tumour.
Choroid Plexus Papilloma
Tumours derived from choroid plexus epithelium are uncommon intracranial tumours. They are found in the distribution of the choroid plexus. In children, they occur most frequently in the lateral ventricles, whereas in adults fourth ventricle is the most common site. They are invariably benign tumours and rarely ever undergo malignant transformation.
MORPHOLOGIC FEATURES. Grossly, the tumour projects as rounded, papillary mass into one of the ventricles.
Histologically, choroid plexus papilloma is a papillary tumour resembling normal choroid plexus with a vascular connective tissue core covered by a single layer of cuboidal epithelium which lies upon a basement membrane.
POORLY-DIFFERENTIATED AND
EMBRYONAL TUMOURS
CNS tumours composed of primitive undifferentiated cells include medulloblastoma and glioblastoma, and rarely, neuroblastoma (page 800) and retinoblastoma (page 512). Except for medulloblastoma, other examples of these tumours have been described elsewhere in the text.
Medulloblastoma
Medulloblastoma is the most common variety of primitive neuroectodermal tumour. It comprises 25% of all childhood brain tumours but a quarter of cases occur in patients over the age of 20 years. The most common location is the cerebellum in the region of roof of fourth ventricle, in the midline of cerebellum, in the vermis, and in the cerebellar hemispheres. Medulloblastoma is a highly malignant tumour and spreads to local as well as to distant sites. It invades locally and by the CSF to meninges, ventricles and subarachnoid space and has a tendency for widespread
metastases to extraneural sites such as to lungs, liver, 889 vertebrae and pelvis.
MORPHOLOGIC FEATURES. Grossly, the tumour typically protrudes into the fourth ventricle as a soft, greywhite mass or invades the surface of the cerebellum.
Microscopically, medulloblastoma is composed of small, poorly-differentiated cells with ill-defined cytoplasmic processes and a tendency to be arranged around blood vessels and occasionally forms pseudorosettes (HomerWright rosettes). Another characteristic of the tumour is differentiation into glial or neuronal elements.
OTHER PRIMARY INTRAPARENCHYMAL TUMOURS
Important examples of some other primary |
intraparen- |
|
||
chymal are haemangioblastoma, CNS lymphoma and germ |
|
|||
cell tumours. |
|
|
|
|
Haemangioblastoma |
|
|
|
|
Haemangioblastoma is a tumour of uncertain origin and |
|
|||
constitutes about 2% of all intracranial tumours. It is seen |
CHAPTER |
|||
more commonly in young adults and is commoner in males. |
||||
|
||||
It may occur sporadically or be a part of von Hippel-Lindau |
|
|||
syndrome (along with cysts in the liver, kidney, and benign/ |
|
|||
malignant renal tumour). About a quarter of haemangio- |
|
|||
blastomas secrete erythropoietin and cause polycythaemia. |
30 |
|||
|
|
|
||
MORPHOLOGIC FEATURES. Grossly, the tumour is |
|
|||
|
|
|||
usually a circumscribed cystic mass with a mural nodule. |
|
The |
||
The cyst contains haemorrhagic fluid. |
|
|
||
|
|
|
||
Microscopically, the features are as under: |
|
|
Nervous |
|
lipid-laden foamy stromal cells. |
|
|
||
i) Large number of thin-walled blood vessels lined by |
|
|
||
plump endothelium. |
|
|
|
|
ii) Vascular spaces are separated by groups of polygonal |
|
|
||
|
|
|
System |
|
Primary CNS Lymphoma |
|
|
||
|
|
|
||
Lymphomas in the brain may occur as a part of disseminated non-Hodgkin’s lymphoma (Chapter 14) or may be a primary CNS lymphoma. The incidence of the primary CNS lymphoma has shown a rising trend in patients of AIDS and other immunosuppressed conditions. They occur in men above 5th decade of life. Primary CNS lymphoma has a poor prognosis.
MORPHOLOGIC FEATURES. Grossly, the tumour is frequently periventricular in location and may appear nodular or diffuse.
Microscopically, the features are as under:
i)Characteristically, the tumour grows around blood vessels i.e. has an angiocentric growth pattern. Reticulin stain highlights this feature well.
ii)Typically, CNS lymphomas are diffuse, large cell type with high mitotic activity.
iii)They are generally β-cell type.
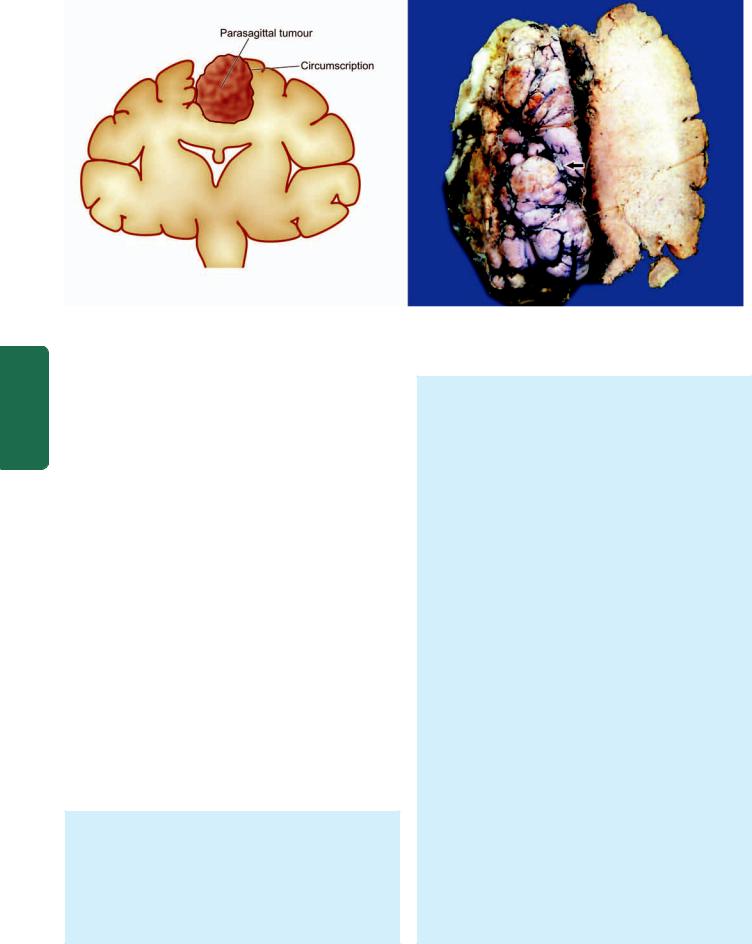
890
Pathology Systemic III SECTION
Figure 30.16 
 Meningioma, The tumour mass is circumscribed (arrow) with irregular surface convolutions and prominent blood vessels. It is firm in consistency with peripherally adherent thick firm dural tissue. Cut surface of the mass is firm and fibrous.
Meningioma, The tumour mass is circumscribed (arrow) with irregular surface convolutions and prominent blood vessels. It is firm in consistency with peripherally adherent thick firm dural tissue. Cut surface of the mass is firm and fibrous.
Germ Cell Tumours
Rarely, germ cell tumours may occur in the brain, especially in children. Common locations are suprasellar region and pineal area. Some common examples of such tumours are germinoma (seminoma/dysgerminoma), teratoma and embryonal carcinoma. Morphologically, they are similar to their counterparts elsewhere.
TUMOURS OF MENINGES
The most common tumour arising from the pia-arachnoid is meningioma accounting for 20% of intracranial tumours.
Meningioma
Meningiomas arise from the cap cell layer of the arachnoid. Their most common sites are in the front half of the head and include: lateral cerebral convexities, midline along the falx cerebri adjacent to the major venous sinuses parasagittally, and olfactory groove. Less frequent sites are: within the cerebral ventricles, foramen magnum, cerebellopontine angle and the spinal cord. Meningiomas are generally solitary. They have an increased frequency in patients with neurofibromatosis 2 and are often multiple in these cases. They are usually found in 2nd to 6th decades of life, with slight female preponderance. Most meningiomas are benign and can be removed successfully. Rarely, a malignant meningioma may metastasise, mainly to the lungs.
MORPHOLOGIC FEATURES. Grossly, meningioma is well-circumscribed, solid, spherical or hemispherical mass of varying size (1-10 cm in diameter). The tumour is generally firmly attached to the dura and indents the surface of the brain but rarely ever invades it (Fig. 30.16). The overlying bone usually shows hyperostosis. Cut surface of the tumour is firm and fibrous, sometimes with foci of calcification.
Microscopically, meningiomas are divided into 5 subtypes: meningotheliomatous (syncytial), fibrous (fibroblastic), transitional (mixed), angioblastic and anaplastic (malignant).
1.Meningotheliomatous (syncytial) meningioma. This pattern of meningioma resembles the normal arachnoid cap cells. The tumour consists of solid masses of polygonal cells with poorly-defined cell membranes (i.e. syncytial appearance). The cells have round to oval, central nuclei with abundant, finely granular cytoplasm. Some amount of collagenous stroma is present that divides the tumour into irregular lobules.
2.Fibrous (fibroblastic) meningioma. A less frequent pattern is of a spindle-shaped fibroblastic tumour in which the tumour cells form parallel or interlacing bundles. Whorled pattern and psammoma bodies are less common features of this type.
3.Transitional (mixed) meningioma. This pattern is characterised by a combination of cells with syncytial and fibroblastic features with conspicuous whorled pattern of tumour cells, often around central capillary-sized blood vessels. Some of the whorls contain psammoma bodies due to calcification of the central core of whorls (Fig. 30.17). Other forms of degenerative changes like xanthomatous and myxomatous degeneration may also be encountered, in transitional variety.
These first three histologic patterns constitute a spectrum of lesions rather than three distinct entities.
4.Angioblastic meningioma. An angioblastic meningioma includes 2 patterns: haemangioblastic pattern resembling haemangioblastoma of the cerebellum, and haemangiopericytic pattern which is indistinguishable from haemangiopericytoma elsewhere in the body. Both types of angioblastic meningiomas have high rate of recurrences.
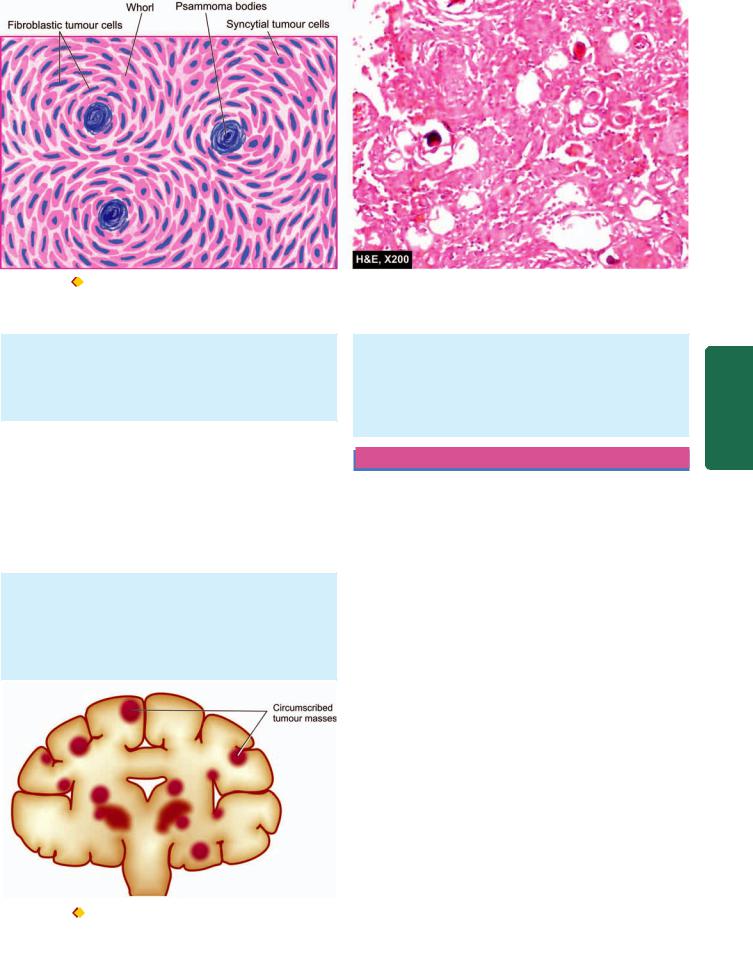
891
Figure 30.17 |
Meningioma, transitional type. The cells have features of both syncytial and fibroblastic type and form whorled appearance. |
Some of the whorls contain psammoma bodies.
5. Anaplastic (malignant) meningioma. Rarely, a meningioma may display features of anaplasia and invade the underlying brain or spinal cord. This pattern of meningioma is associated with extraneural metastases, mainly to the lungs.
METASTATIC TUMOURS
brain and spinal cord, particularly encountered in carcinomas of the lung and breast.
Histologically, metastatic tumours in the brain recapitulate the appearance of the primary tumour of origin with sharp line of demarcation from adjoining brain tissue. It is usually surrounded by a zone of oedema.
Approximately a quarter of intracranial tumours are metastatic tumours. The clinical features are like those of a primary brain tumour. Most common primary tumours metastasising to the brain are: carcinomas of the lung, breast, skin (malignant melanoma), kidney and the gastrointestinal tract and choriocarcinoma. Infiltration from lymphoma and leukaemias may also occur.
MORPHOLOGIC FEATURES. Grossly, the metastatic deposits in the brain are usually multiple, sharply-defined masses at the junction of grey and white matter (Fig. 30.18). A less frequent pattern is carcinomatous meningitis or meningeal carcinomatosis in which there is presence of carcinomatous nodules on the surface of the
Figure 30.18 |
Metastatic tumour deposits in the brain. They are |
commonly multiple, well-defined and usually located at the grey and white matter junction.
PERIPHERAL NERVOUS SYSTEM
NORMAL STRUCTURE
The peripheral nervous system (PNS) consists of cranial and spinal nerves, sympathetic and parasympathetic autonomic nervous system and the peripheral ganglia. The PNS is involved in electric transmission of sensory and motor impulses to and from the CNS. A peripheral nerve is surrounded by an outer layer of fibrous tissue, the epineurium. Each nerve is made of several fascicles enclosed in multilayered membrane of flattened cells, the perineurium. Each fascicle is composed of bundles of connective tissue, the endoneurium. There are 2 main types of nerve fibres or axons comprising a peripheral nerve—myelinated and nonmyelinated. Myelinated axons are thicker (diameter greater than 2 μm) and are surrounded by a chain of Schwann cells which produce myelin sheath. Non-myelinated axons have diameter of 0.2-3 μm and about ten non-myelinated fibres may be enclosed by a Schwann cell. Nodes of Ranvier on myelinated fibres are the boundaries between each Schwann cell surrounding the fibre (Fig. 30.18). Myelinated axons have their origin from neurons in the posterior root ganglia and the anterior horn cell of the spinal cord, whereas nonmyelinated axons arise from neurons in the posterior root ganglia and in the autonomic ganglia.
PATHOLOGIC REACTIONS TO INJURY
The peripheral nerves, unlike brain, have regenerative capacity as has been discussed on page 172. The pathologic reactions of the PNS in response to injury may be in the
System Nervous The 30 CHAPTER
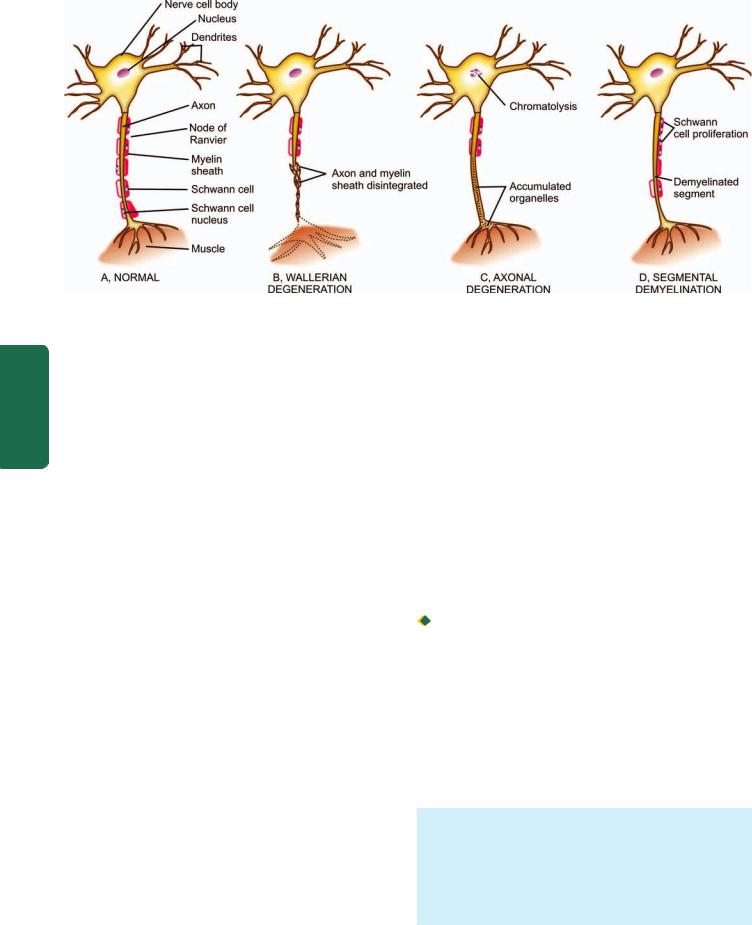
892
Pathology Systemic III SECTION
Figure 30.19 
 Pathologic reaction of peripheral nerve to injury.
Pathologic reaction of peripheral nerve to injury.
form of one of the types of degenerations causing peripheral neuropathy or formation of a traumatic neuroma. There are 3 main types of degenerative processes in the PNS— Wallerian degeneration, axonal degeneration and segmental demyelination (Fig. 30.19):
WALLERIAN DEGENERATION. Wallerian degeneration occurs after transection of the axon which may be as a result of knife wounds, compression, traction and ischaemia. Following transection, initially there is accumulation of organelles in the proximal and distal ends of the transection sites. Subsequently, the axon and myelin sheath distal to the transection site undergo disintegration upto the next node of Ranvier, followed by phagocytosis. The process of regeneration occurs by sprouting of axons and proliferation of Schwann cells from the proximal end.
AXONAL DEGENERATION. In axonal degeneration, degeneration of the axon begins at the peripheral terminal and proceeds backward towards the nerve cell body. The cell body often undergoes chromatolysis. There is Schwann cell proliferation in the region of axonal degeneration. The loss of axonal integrity occurs, probably as a result of some primary metabolic disturbance within the axon itself. Changes similar to those seen in Wallerian degeneration are present but regenerative reaction is limited or absent.
SEGMENTAL DEMYELINATION. Segmental demyelination is similar to demyelination within the brain (page 884). Segmental demyelination is loss of myelin of the segment between two consecutive nodes of Ranvier, leaving a denuded axon segment. The axon, however, remains intact. Schwann cell proliferation generally accompanies demyelination. This results in remyelination of the affected axon. Repeated episodes of demyelination and remyelination are associated with concentric proliferation of Schwann cells
around axons producing ‘onion bulbs’ found in hypertrophic neuropathy.
TRAUMATIC NEUROMA. Normally, the injured axon of a peripheral nerve regenerates at the rate of approximately 1 mm per day. However, if the process of regeneration is hampered due to an interposed haematoma or fibrous scar, the axonal sprouts together with Schwann cells and fibroblasts form a peripheral mass called as traumatic or stump neuroma.
PERIPHERAL NEUROPATHY
Peripheral neuropathy is the term used for disorders of the peripheral nerves of any cause. It may be polyneuropathy, mononeuropathy multiplex, and mononeuropathy.

 Polyneuropathy is characteristically symmetrical with noticeable sensory features such as tingling, pricking, burning sensation or dysaesthesia in feet and toes. Motor features in the form of muscle weakness and loss of tendon reflexes may be present. Involvement of the autonomic nervous system may be associated. Most cases have origin in acquired metabolic and toxic causes such as thiamine deficiency, diabetes, amyloidosis, autoimmune demyelinating disease (Guillain-Barré syndrome), and administration of toxins and certain therapeutic agents (e.g. vincristine, isoniazid). Besides these, a number of hereditary polyneuropathies are described.
Polyneuropathy is characteristically symmetrical with noticeable sensory features such as tingling, pricking, burning sensation or dysaesthesia in feet and toes. Motor features in the form of muscle weakness and loss of tendon reflexes may be present. Involvement of the autonomic nervous system may be associated. Most cases have origin in acquired metabolic and toxic causes such as thiamine deficiency, diabetes, amyloidosis, autoimmune demyelinating disease (Guillain-Barré syndrome), and administration of toxins and certain therapeutic agents (e.g. vincristine, isoniazid). Besides these, a number of hereditary polyneuropathies are described.
Pathologically, polyneuropathy may be the result of axonal degeneration (axonopathy) or segmental demyelination (demyelinating polyneuropathy). In each type, acute, subacute and chronic forms are distinguished. Guillain-Barré syndrome is the classical example of acute demyelinating polyneuropathy which has probably an autoimmune etiology.
893
Figure 30.20 
 Schwannoma (neurilemmoma), showing whorls of densely cellular (Antoni A) and loosely cellular (Antoni B) areas with characteristic nuclear palisading (Verocay bodies).
Schwannoma (neurilemmoma), showing whorls of densely cellular (Antoni A) and loosely cellular (Antoni B) areas with characteristic nuclear palisading (Verocay bodies).

 Mononeuropathy multiplex or multifocal neuropathy is defined as simultaneous or sequential multifocal involvement of nerve trunks which are not in continuity. The involvement may be partial or complete and may evolve over days or years. Multifocal neuropathy represents part of spectrum of chronic acquired demyelinating neuropathy.
Mononeuropathy multiplex or multifocal neuropathy is defined as simultaneous or sequential multifocal involvement of nerve trunks which are not in continuity. The involvement may be partial or complete and may evolve over days or years. Multifocal neuropathy represents part of spectrum of chronic acquired demyelinating neuropathy.

 Mononeuropathy, on the other hand, is focal involvement of a single nerve. It is generally the result of local causes such as direct trauma, compression and entrapment.
Mononeuropathy, on the other hand, is focal involvement of a single nerve. It is generally the result of local causes such as direct trauma, compression and entrapment.
NERVE SHEATH TUMOURS
Tumours of the peripheral nerves are commonly benign and include schwannoma (neurilemmoma) and neurofibroma. Both of them arise from Schwann cells but neurofibroma contains large amount of collagen. Rarely, their malignant counterpart, malignant peripheral nerve sheath tumour, develops particularly in patients with von Recklinghausen’s neurofibromatosis.
Schwannomas (Neurilemmomas)
Schwannomas or neurilemmomas arise from cranial and spinal nerve roots. An acoustic schwannoma or acoustic neuroma is an intracranial schwannoma located within the internal auditory canal originating from vestibular portion of the acoustic nerve (page 515). Intraspinal schwannomas are found as intradural tumours in the thoracic region. In the peripheral nerves, they occur as solitary nodule on any sheathed sensory, motor, or autonomic nerve. Multiple schwannomas are uncommon and occur in von Recklinghausen’s disease (see below). Schwannomas are tumours of adults except in von Recklinghausen’s disease.
MORPHOLOGIC FEATURES. Grossly, a schwannoma is an encapsulated, solid, sometimes cystic, tumour that produces eccentric enlargement of the nerve root from where it arises.
Microscopically, the tumour is composed of fibrocellular bundles forming whorled pattern. There are areas of dense and compact cellularity (Antoni A pattern) alternating with loose acellular areas (Antoni B pattern). Areas of Antoni A pattern show palisaded nuclei called Verocay bodies (Fig. 30.20). Nerve fibres are usually found stretched over the capsule but not within the tumour. Areas of degeneration contain haemosiderin and lipid-laden macrophages. Schwann cells characteristically express S-100 protein. A schwannoma rarely ever becomes malignant.
Neurofibromas and von Recklinghausen’s Disease
Neurofibromas may occur as solitary, fusiform cutaneous tumour of a single nerve, but more often are multiple associated with von Recklinghausen’s disease. Solitary neurofibroma is a tumour of adults but multiple neurofibromas or neurofibromatosis is a hereditary disorder with autosomal dominant inheritance. Solitary neurofibroma is generally asymptomatic but patients with von Recklinghausen’s disease have a triad of features:

 Multiple cutaneous neurofibromas.
Multiple cutaneous neurofibromas.

 Numerous pigmented skin lesions (‘cafe au lait’ spots).
Numerous pigmented skin lesions (‘cafe au lait’ spots).

 Pigmented iris hamartomas.
Pigmented iris hamartomas.
Neurofibromatosis type 1 is a genetic disorder having mutation in chromosome 17 while type 2 has mutation in chromosome 22.
MORPHOLOGIC FEATURES. Grossly, neurofibroma is an unencapsulated tumour producing fusiform enlargement of the affected nerve. Neurofibromatosis in von Recklinghausen’s disease is characterised by numerous nodules of varying size, seen along the small cutaneous nerves but may also be found in visceral branches of sympathetic nerves. Neurofibromatosis may involve a
System Nervous The 30 CHAPTER
894
|
Figure 30.21 |
Plexiform neurofibromatosis. The main mass is |
|
|
multilobulated with increased fat while lower part of the image shows a |
||
|
separate encapsulated gelatinous mass. Cut surface of both the masses |
||
SECTION |
shows circumscribed, gelatinous, lobulated grey-white firm masses. |
||
|
|||
neurofibroma) (Fig. 30.21). |
|||
|
group of nerves or may occur as multiple, oval and |
||
|
irregular swellings along the length of a nerve (plexiform |
||
III |
Microscopically, a neurofibroma is composed of bundles |
||
and interlacing fascicles of delicate and elongated spindle- |
|||
|
|||
|
shaped cells having wavy nuclei. The cellular area is |
||
Systemic |
separated by loose collagen and mucoid material. Residual |
||
nerve fibres (neurites) may be demonstrable (Fig. 30.22). |
|||
|
|||
|
Histologic appearance of Antoni B pattern of schwannoma |
||
|
may be seen in neurofibroma and cause diagnostic |
||
Pathology |
difficulty. Immunohistochemically, neurofibroma is |
||
positive for epithelial membrane antigen (EMA) and some |
|||
|
|||
|
tumours express S-100 protein as schwannomas do. |
||
|
|
|
|
Neurofibromas have tendency for local recurrences after excision. Neurilemmoma virtually never turns malignant, while sarcomatous transformation in neurofibroma, particularly in neurofibromatosis, is not unusual. It is estimated that about 3% of patients with von Recklinghausen’s neurofibromatosis develop malignant transformation of one of the nodules. Rarely, neurogenic sarcoma may develop spontaneously in the absence of preexisting von Recklinghausen’s disease.
The contrasting features to distinguish neurofibroma from schwannoma are listed in Table 30.5.
Malignant Peripheral Nerve Sheath Tumour
Malignant peripheral nerve sheath tumour (MPNST) is a poorly differentiated spindle cell sarcoma of the peripheral nerves occurring most often in adults. The tumour may arise de novo or from malignant transformation of a pre-existing neurofibroma than a schwannoma, generally at an early age (20-40 years). About 50% of the tumours are seen in patients with neurofibromatosis type 1 with chromosomal deletion 17p and p53 gene mutations, while some develop at sites of previous irradiation.
MORPHOLOGIC FEATURES. Grossly, the tumour appears as an unencapsulated fusiform enlargement of a nerve.
Microscopically, the tumour has the general appearance of tumour cells resembling a fibrosarcoma. The tumour has frequent mitosis and areas of necrosis. Triton tumour is the name used for MPNST which has areas of poorlydifferentiated rhabdomyosarcoma, cartilage and bone.
Epithelioid MPNST has plump cells resembling epithelioid cells and is positive for HMB-45 immunostain. Most of the recurrent forms of MPNST are of epithelioid type.
Figure 30.22 
 Neurofibroma, showing interlacing bundles of spindle-shaped cells separated by mucoid matrix. The cells have wavy nuclei and a residual nerve fibre (neurite) is also identified.
Neurofibroma, showing interlacing bundles of spindle-shaped cells separated by mucoid matrix. The cells have wavy nuclei and a residual nerve fibre (neurite) is also identified.
TABLE 30.5: Contrasting Features of Neurilemmoma (Schwannoma) and Neurofibroma. |
895 |
||
Feature |
Neurilemmoma |
Neurofibroma |
|
|
|
|
|
1. Location |
Cerebellopontine angle (vestibular branch of |
Dermis; along the nerve trunk |
|
|
8th nerve); extradural sites |
|
|
2. |
Number |
Generally solitary |
3. |
Genetics |
Bilateral in association with type 2 neuro- |
|
|
fibromatosis having autosomal dominant |
|
|
inheritance (chromosome 22 disorder) |
4. |
Gross appearance |
Firm, encapsulated, |
|
|
c/s tan, translucent |
5. |
Microscopy |
Compact areas (Antoni A) and myxomatous |
|
|
areas (Antoni B), palisading tumour cells |
|
|
(Verocay bodies) |
Solitary or multiple neurofibromatosis
Multiple associated with type 1 neurofibromatosis having autosomal dominant inheritance (chromosome 17 disorder)
Soft, well demarcated but unencapsulated, c/s mucoid, translucent
Dense collagen fibres and abundant extracellular mucoid material
6. |
Infiltration |
Encapsulated along the edge of |
May infiltrate the peripheral nerve |
|
|
nerve without invading it |
|
7. |
Immunohistochemistry |
S-100 protein |
EMA; sometimes S-100 protein |
8. |
Behaviour |
Invariably benign |
May turn malignant |
|
|
|
|
Although relatively slow-growing, MPNST has local recurrences and haematogenous metastasis occur
commonly; histology thus appears to have little correlation with clinical behaviour of the tumour.
System Nervous The 30 CHAPTER