

- •Foreword
- •Preface
- •Contents
- •1. Introduction to Pathology
- •2. Techniques for the Study of Pathology
- •6. Inflammation and Healing
- •8. Neoplasia
- •16. The Heart
- •17. The Respiratory System
- •18. The Eye, ENT and Neck
- •20. The Gastrointestinal Tract
- •24. The Female Genital Tract
- •25. The Breast
- •26. The Skin
- •27. The Endocrine System
- •28. The Musculoskeletal System
- •29. Soft Tissue Tumours
- •30. The Nervous System
- •Appendix
- •Further Readings
- •Index
791
Chapter 27 |
The Endocrine System |
ENDOCRINES: THE BASIC CONCEPT
The development, structure and functions of human body are governed and maintained by 2 mutually interlinked systems—the endocrine system and the nervous system (Chapter 30); a third system combining features of both these systems is appropriately called neuroendocrine system.
NEUROENDOCRINE SYSTEM
This system forms a link between the endocrine and nervous systems. The cells of this system elaborate polypeptide hormones; owing to these biochemical properties, it has also been called as APUD cell system (acronym for Amine Precursor Uptake and Decarboxylation properties). However, though having common biochemical properties, the cells of this system are widely distributed in the body in different anatomic areas and hence is currently called dispersed neuroendocrine system. Cells comprising this system are as under:
1.Neuroendocrine cells are present in the gastric and intestinal mucosa and elaborate peptide hormones.
2.Neuroganglia cells lie in the ganglia cells in the sympathetic chain and elaborate amines.
3.Adrenal medulla elaborates epinephrine and norepinephrine.
4.Parafollicular C cells of the thyroid secrete calcitonin.
5.Islets of Langerhans in the pancreas (included in both endocrine and neuroendocrine systems) secrete insulin.
6.Isolated cells in the left atrium of the heart secrete atrial natriuretic (salt-losing) peptide hormone.
In addition to above, other non-endocrine secretions include neurotransmitter substances such as acetylcholine and dopamine released from neural synapses, and erythropoietin and vitamin D3 elaborated from the kidney.
THE ENDOCRINE SYSTEM
Anatomically, the endocrine system consists of 6 distinct organs: pituitary, adrenals, thyroid, parathyroids, gonads, and pancreatic islets; the last one is included in neuroendocrine system also). Understanding the patholgy of these endocrine organs requires the knowledge of overall framework of hormone secretions, their actions and broad principles of feedback mechanisms.
Broadly speaking, human hormones are divided into 5 major classes which are further grouped under two headings depending upon their site of interactions on the target cell receptors (whether cell membrane or nuclear receptor):
Group I: Those interacting with cell-surface membrane receptors:
1.Amino acid derivatives: thyroid hormone, catecholamines.
2.Small neuropeptides: gonadotropin-releasing hormone (GnRH), thyrotropin-releasing hormone (TRH), somatostatin, vasopressin.
Group II: Those interacting with intracellular nuclear receptors:
3.Large proteins: insulin, luteinising hormone (LH), parathormone hormone.
4.Steroid hormones: cortisol, estrogen.
5.Vitamin derivatives: retinol (vitamin A) and vitamin D. The synthesis of these hormones and their precursors takes
place through a prescribed genetic pathway that involves: transcription → mRNA → protein synthesis → posttranslational protein processing → intracellular sorting/ membrane integration → secretion.
Major functions of hormones are as under:
i)Growth and differentiation of cells: by pituitary hormones, thyroid, parathyroid, steroid hormones.
ii)Maintenance of homeostasis: thyroid (by regulating BMR), parathormone, mineralocorticoids, vasopressin, insulin.
iii)Reproduction: sexual development and activity, pregnancy, foetal development, menopause etc.
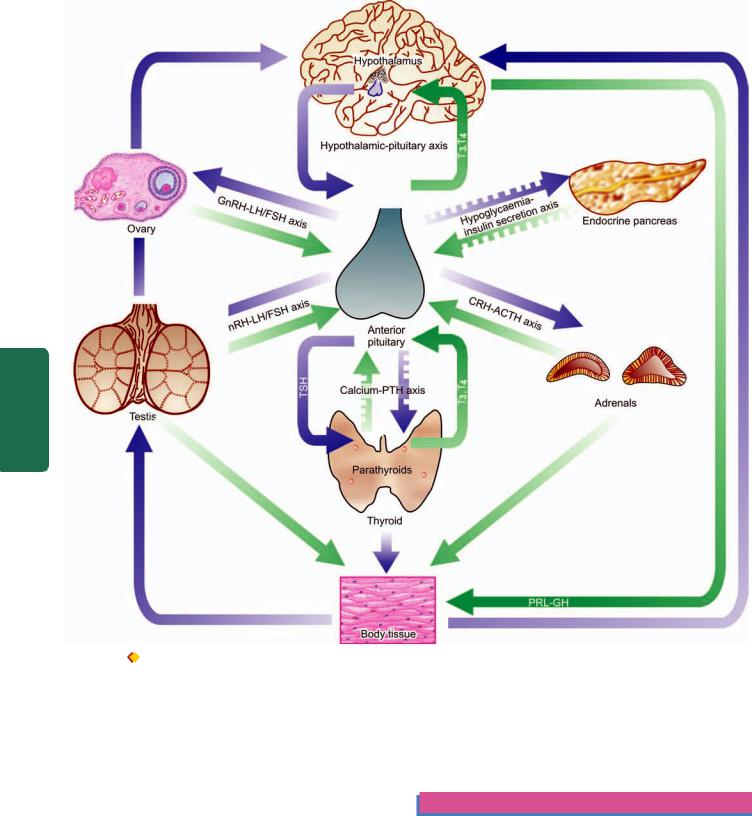
A basic feature of all endocrine glands is the existence of both negative and positive feedback control system that stimulates or regulates hormone production in a way that levels remain within the normal range (abbreviated as S or R respectively with the corresponding hormone e.g. TSH-TRH pathway, GnRH-LH/FSH pathway etc). This system is commonly termed hypothalamic-pituitary hormone axis for different hormones schematically illustrated in Fig. 27.1. The stimulatory or regulatory action by endocrine hormonal secretions may follow paracrine or autocrine pathways:

 Paracrine regulation means that the stimulatory/ regulatory factors are released by one type of cells but act on another adjacent cell of the system.
Paracrine regulation means that the stimulatory/ regulatory factors are released by one type of cells but act on another adjacent cell of the system.
 Autocrine regulation refers to action of the factor on the same cell that produced it.
Autocrine regulation refers to action of the factor on the same cell that produced it.
With this brief overview of principles of physiology of hormones, we now turn to the study of diseases of the endocrine organs. In general, pathologic processes affecting endocrine glands with resultant hormonal abnormalities may occur from following processes:
Hyperfunction: This results from excess of hormone secreting tissues e.g. hyperplasia, tumours (adenoma, carcinoma), ectopic hormone production, excessive stimulation from
System Endocrine The 27 CHAPTER
792
Pathology Systemic III SECTION
Figure 27.1 
 Endocrine organs and the presence of feedback controls. Both positive and negative feedback controls exist for each endocrine gland having a regulating (R) and stimulating (S) hormone. Those acting through hypothalamic-pituitary axis include: thyroid hormones on TRH-
Endocrine organs and the presence of feedback controls. Both positive and negative feedback controls exist for each endocrine gland having a regulating (R) and stimulating (S) hormone. Those acting through hypothalamic-pituitary axis include: thyroid hormones on TRH-
TSH axis, cortisol on CRH-ACTH axis, gonadal steroids on GnRH-LH/FSH axis and insulin-like GH on GHRH-GH axis. Those independent of pituitary control (shown by interrupted arrows) have also feedback controls by calcium on PTH, and hypoglycaemia on insulin release by pancreatic islets.
inflammation (often autoimmune), infections, iatrogenic (drugs-induced, hormonal administration).
Hypofunction: Deficiency of hormones occurs from destruction of hormone-forming tissues from inflammations (often autoimmune), infections, iatrogenic (e.g. surgical removal, radiation damage), developmental defects (e.g. Turner’s syndrome, hypoplasia), enzyme deficiency, haemorrhage and infarction (e.g. Sheehan’s syndrome), nutritional deficiency (e.g. iodine deficiency).
Hormone resistance: There may be adequate or excessive production of a hormone but there is peripheral resistance, often from inherited mutations in receptors (e.g. defect in
membrane receptors, nuclear receptors or receptor for signal transduction).
PITUITARY GLAND
NORMAL STRUCTURE
ANATOMY. The pituitary gland or hypophysis in an adult weighs about 500 mg and is slightly heavier in females. It is situated at the base of the brain in a hollow called sella turcica formed out of the sphenoid bone. The gland is composed of 2 major anatomic divisions: anterior lobe (adenohypophysis) and posterior lobe (neurohypophysis).

1.The anterior lobe or adenohypophysis is an ectodermal derivative formed from Rathke’s pouch which is an upward diverticulum from the primitive buccal cavity. The adenohypophysis has no direct neural connection but has indirect connection through capillary portal circulation by which the anterior pituitary receives the blood which has already passed through the hypothalamus.
2.The posterior lobe or neurohypophysis is a downgrowth from the primitive neural tissue. The neurohypophysis, therefore, has direct neural connection superiorly with the hypothalamus.
HISTOLOGY AND FUNCTIONS. The histology and functions of the anterior and posterior lobes of the pituitary gland are quite distinct.
A. ANTERIOR LOBE (ADENOHYPOPHYSIS). It is composed of round to polygonal epithelial cells arranged in cords and islands having fibrovascular stroma. These epithelial cells, depending upon their staining characteristics and functions, are divided into 3 types, each of which performs separate functions:
1. Chromophil cells with acidophilic granules: These cells comprise about 40% of the anterior lobe and are chiefly located in the lateral wings. The acidophils are further of 2 types:
i)Somatotrophs (GH cells) which produce growth hormone (GH).
ii)Lactotrophs (PRL cells) which produce prolactin (PRL). Cells containing both GH and PRL called mammo-
somatotrophs are also present.
2. Chromophil cells with basophilic granules: These cells constitute about 10% of the anterior lobe and are mainly found in the region of median wedge. The chromatophils include 3 types of cells:
i)Gonadotrophs (FSH-LH cells) which are the source of the FSH and LH or interstitial cell stimulating hormone (ICSH).
ii)Thyrotrophs (TSH cells) are the cells producing TSH.
iii)Corticotrophs (ACTH-MSH cells) produce ACTH, melanocyte stimulating hormone (MSH), β-lipoprotein and β-endorphin.
3. Chromophobe cells without visible granules: These cells comprise the remainder 50% of the adenohypophysis. These cells by light microscopy contain no visible granules, but on electron microscopy reveal sparsely granulated corticotrophs, thyrotrophs and gonadotrophs.
All these functions of the adenohypophysis are under the indirect control of the hypothalamus through stimulatory and inhibitory factors synthesised by the hypothalamus which reach the anterior lobe through capillary portal blood.
B. POSTERIOR LOBE (NEUROHYPOPHYSIS). The neurohypophysis is composed mainly of interlacing nerve fibres in which are scattered specialised glial cells called pituicytes. These nerve fibres on electron microscopy contain granules of neurosecretory material made up of 2 octapeptides—vasopressin or antidiuretic hormone (ADH), and oxytocin, both of which are produced by neurosecretory cells
of the hypothalamus but are stored in the cells of posterior pituitary.
1.ADH causes reabsorption of water from the renal tubules and is essential for maintenance of osmolality of the plasma. Its deficiency results in diabetes insipidus characterised by uncontrolled diuresis and polydipsia.
2.Oxytocin causes contraction of mammary myoepithelial cells resulting in ejection of milk from the lactating breast and causes contraction of myometrium of the uterus at term.
It is obvious from the description above that pituitary, though a tiny organ, is concerned with a variety of diverse functions in the body. The pituitary gland and hypothalamus are so closely interlinked that diseases of the pituitary gland involve the hypothalamus, and dysfunctions of the hypothalamus cause secondary changes in the pituitary. The pituitary gland is involved in several diseases which include: non-neoplastic (e.g. inflammations, haemorrhage, trauma, infarction and many other endocrine diseases) and neoplastic diseases. However, functionally and morphologically, diseases of the pituitary can be classified as below, each of which includes diseases of the anterior and posterior pituitary and the hypothalamus, separately:
i)Hyperpituitarism
ii)Hypopituitarism
iii)Pituitary tumours
HYPERPITUITARISM
Hyperpituitarism is characterised by oversecretion of one or more of the pituitary hormones. Such hypersecretion may be due to diseases of the anterior pituitary, posterior pituitary or hypothalamus. For all practical purposes, however, hyperfunction of the anterior pituitary is due to the development of a hormone-secreting pituitary adenoma (discussed later), and rarely, a carcinoma. For each of the hormonal hyperfunction of the anterior pituitary, posterior pituitary and hypothalamus, a clinical syndrome is described. A few important syndromes are as follows:
A. Hyperfunction of Anterior Pituitary
Three common syndromes of adenohypophyseal hyperfunction are: gigantism and acromegaly, hyperprolactinaemia and Cushing’s syndrome.
GIGANTISM AND ACROMEGALY. Both these clinical syndromes result from sustained excess of growth hormone (GH), most commonly by somatotroph (GH-secreting) adenoma.
Gigantism. When GH excess occurs prior to epiphyseal closure, gigantism is produced. Gigantism, therefore, occurs in prepubertal boys and girls and is much less frequent than acromegaly. The main clinical feature in gigantism is the excessive and proportionate growth of the child. There is enlargement as well as thickening of the bones resulting in considerable increase in height and enlarged thoracic cage.
Acromegaly. Acromegaly results when there is overproduction of GH in adults following cessation of bone growth and
793
System Endocrine The 27 CHAPTER

794 is more common than gigantism. The term ‘acromegaly’ means increased growth of extremities (acro=extremity). There is enlargement of hands and feet, coarseness of facial features with increase in soft tissues, prominent supraorbital ridges and a more prominent lower jaw which when clenched results in protrusion of the lower teeth in front of upper teeth (prognathism). Other features include enlargement of the tongue and lips, thickening of the skin and kyphosis. Sometimes, a few associated features such as TSH excess resulting in thyrotoxicosis, and gonadotropin insufficiency causing amenorrhoea in the females and impotence in the male, are found.
|
HYPERPROLACTINAEMIA. Hyperprolactinaemia is the |
|
|
excessive production of prolactin (PRL), most commonly by |
|
|
lactotroph (PRL-secreting) adenoma, also called prolac- |
|
|
tinoma. Occasionally, hyperprolactinaemia results from |
|
|
hypothalamic inhibition of PRL secretion by certain drugs |
|
|
(e.g. chlorpromazine, reserpine and methyl-dopa). In the |
|
|
female, hyperprolactinaemia causes amenorrhoea-galactorrhoea |
|
|
syndrome characterised clinically by infertility and expression |
|
|
of a drop or two of milk from breast, not related to pregnancy |
|
SECTION |
or puerperium. In the male, it may cause impotence or reduced |
|
libido. These features result either from associated inhibition |
||
|
||
|
of gonadotropin secretion or interference in gonadotropin |
|
|
effects. |
|
|
CUSHING’S SYNDROME. Pituitary-dependent Cushing’s |
|
III |
syndrome results from ACTH excess. Most frequently, it is |
|
caused by corticotroph (ACTH-secreting) adenoma. |
||
|
||
|
Cushing’s syndrome is discussed under diseases of the |
|
Systemic |
adrenal gland on page 797. |
|
B. Hyperfunction of Posterior Pituitary and |
||
|
||
|
Hypothalamus |
|
Pathology |
Lesions of posterior pituitary and hypothalamus are |
|
INAPPROPRIATE RELEASE OF ADH. Inappropriate |
||
|
uncommon. Two of the syndromes associated with hyper- |
|
|
function of the posterior pituitary and hypothalamus are: |
|
|
inappropriate release of ADH and precocious puberty. |
|
|
release of ADH results in its excessive secretion which |
|
|
manifests clinically by passage of concentrated urine due to |
|
|
increased reabsorption of water and loss of sodium in the |
|
|
urine, consequent hyponatraemia, haemodilution and |
|
|
expansion of intraand extracellular fluid volume. |
|
|
Inappropriate release of ADH occurs most often in |
|
|
paraneoplastic syndrome e.g. in oat cell carcinoma of the |
|
|
lung, carcinoma of the pancreas, lymphoma and thymoma. |
|
|
Infrequently, lesions of the hypothalamus such as trauma, |
|
|
haemorrhage and meningitis may produce ADH |
|
|
hypersecretion. Rarely, pulmonary diseases such as |
|
|
tuberculosis, lung abscess, pneumoconiosis, empyema and |
|
|
pneumonia may cause overproduction of ADH. |
|
|
PRECOCIOUS PUBERTY. A tumour in the region of |
|
|
hypothalamus or the pineal gland may result in premature |
|
|
release of gonadotropins causing the onset of pubertal |
|
|
changes prior to the age of 9 years. The features include |
|
|
premature development of genitalia both in the male and in |
the female, growth of pubic hair and axillary hair. In the female, there is breast growth and onset of menstruation.
HYPOPITUITARISM
In hypopituitarism, there is usually deficiency of one or more of the pituitary hormones affecting either anterior pituitary, or posterior pituitary and hypothalamus.
A. Hypofunction of Anterior Pituitary
Adenohypophyseal hypofunction is invariably due to destruction of the anterior lobe of more than 75% because the anterior pituitary possesses a large functional reserve. This may result from anterior pituitary lesions or pressure and destruction from adjacent lesions. Lesions of the anterior pituitary include nonsecretory (chromophobe) adenoma, metastatic carcinoma, craniopharyngioma, trauma, postpartum ischaemic necrosis (Sheehan’s syndrome), empty-sella syndrome, and rarely, tuberculosis. Though a number of syndromes associated with deficiency of anterior pituitary hormones have been described, two important syndromes are panhypopituitarism and dwarfism.
PANHYPOPITUITARISM. The classical clinical condition of major anterior pituitary insufficiency is called panhypopituitarism. Three most common causes of panhypopituitarism are: non-secretory (chromophobe) adenoma (discussed later), Sheehan’s syndrome and Simmond’s disease, and empty-sella syndrome.
Sheehan’s syndrome and Simmond’s disease. Pituitary insufficiency occurring due to postpartum pituitary (Sheehan’s) necrosis is called Sheehan’s syndrome, whereas occurrence of similar process without preceding pregnancy as well as its occurrence in males is termed Simmond’s disease. The main pathogenetic mechanism underlying Sheehan’s necrosis is the enlargement of the pituitary occurring during pregnancy which may be followed by hypotensive shock precipitating ischaemic necrosis of the pituitary. Other mechanisms hypothesised are: DIC following delivery, traumatic injury to vessels, and excessive haemorrhage. Patients with long-standing diabetes mellitus appear to be at greater risk of developing this complication.
The first clinical manifestation of Sheehan’s syndrome is failure of lactation following delivery which is due to deficiency of prolactin. Subsequently, other symptoms develop which include loss of axillary and pubic hair, amenorrhoea, sterility and loss of libido. Concomitant deficiency of TSH and ACTH may result in hypothyroidism and adrenocortical insufficiency.
The morphologic features in the anterior pituitary in Sheehan’s syndrome during early stage are ischaemic necrosis and haemorrhage, while later necrotic tissue is replaced by fibrous tissue.
Empty-sella syndrome. Empty-sella syndrome is characterised by the appearance of an empty sella and features of panhypopituitarism. Most commonly, it results from herniation of subarachnoid space into the sella turcica due
to an incomplete diaphragma sella creating an empty sella. Other less common causes are Sheehan’s syndrome, infarction and scarring in an adenoma, irradiation damage, or surgical removal of the gland.
PITUITARY DWARFISM. Severe deficiency of GH in children before growth is completed results in retarded growth and pituitary dwarfism. Most commonly, isolated GH deficiency is the result of an inherited autosomal recessive disorder. Less often it may be due to a pituitary adenoma or craniopharyngioma, infarction and trauma to the pituitary. The clinical features of inherited cases of pituitary dwarfism appear after one year of age. These include proportionate retardation in growth of bones, normal mental state for age, poorly-developed genitalia, delayed puberty and episodes of hypoglycaemia. Pituitary dwarf must be distinguished from hypothyroid dwarf (cretinism) in which there is achondroplasia and mental retardation (page 803).
B.Hypofunction of Posterior Pituitary and Hypothalamus
Insufficiency of the posterior pituitary and hypothalamus is uncommon. The only significant clinical syndrome due to hypofunction of the neurohypophysis and hypothalamus is diabetes insipidus.
DIABETES INSIPIDUS. Deficient secretion of ADH causes diabetes insipidus. The causes of ADH deficiency are: inflammatory and neoplastic lesions of the hypothalamohypophyseal axis, destruction of neurohypophysis due to surgery, radiation, head injury, and lastly, are those cases where no definite cause is known and are labelled as idiopathic. The main features of diabetes insipidus are excretion of a very large volume of dilute urine of low specific gravity (below 1.010), polyuria and polydipsia.
PITUITARY TUMOURS
Tumours of the anterior pituitary are more common than those of the posterior pituitary and hypothalamus. The most common of the anterior pituitary tumours are adenomas; primary and metastatic carcinomas being rare. Cranio-
pharyngioma and granular cell tumour (choristoma) are the other benign pituitary tumours found occasionally.
All pituitary tumours, whether benign or malignant, cause symptoms by following 2 ways:
1.Pressure effects. These are caused by expansion of the lesion resulting in destruction of the surrounding glandular tissue by pressure atrophy. This causes erosion and enlargement of sella turcica, upward extension of the tumour damaging the optic chiasma, optic nerves, neurohypophysis and adjacent cranial nerves, and rarely, downward extension into the nasopharynx.
2.Hormonal effects. Depending upon their cell types, pituitary adenomas produce excess of pituitary hormones and the corresponding clinical syndromes of hyperpituitarism. Infarction and destruction of adenoma may cause symptoms of hypopituitarism.
Pituitary Adenomas
Adenomas are the most common pituitary tumours. They are conventionally classified according to their H & E staining characteristics of granules into acidophil, basophil and chromophobe adenomas. However, this morphologic classification is considered quite inadequate because of the significant functional characteristics of each type of adenoma including the chromophobe adenoma, which on H & E staining does not show visible granules. As a result of advances in the ultrastructural and immunocytochemical studies, a functional classification of pituitary adenoma has emerged. Table 27.1 presents a classification of pituitary adenomas based on functional features as correlated with morphologic features of older classification. The syndromes produced by the tumours have been described already.
MORPHOLOGIC FEATURES. Grossly, pituitary adenomas range in size from small foci of less than 10 mm in size (termed microadenoma) to large adenomas several centimeters in diameter. They are spherical, soft and encapsulated.
Histologically, by light microscopy of H & E stained sections, an adenoma is composed predominantly of one of the normal cell types of the anterior pituitary i.e.
795
System Endocrine The 27 CHAPTER
TABLE 27.1: Morphologic and Functional Classification of Pituitary Adenomas.
|
Functional Type |
Frequency |
Hormones Produced |
Clinical Syndrome |
|
|
|
|
|
1. |
Lactotroph adenoma (Prolactinoma) |
20-30% |
PRL |
Hypogonadism, galactorrhoea |
2. |
Somatotroph adenoma |
5% |
GH |
Acromegaly/gigantism |
3. |
Mixed somatotroph-lactotroph adenoma |
5% |
PRL, GH |
Acromegaly, hypogonadism, |
|
|
|
|
galactorrhoea |
4. |
Corticotroph adenoma |
10-15% |
ACTH |
Cushing’s syndrome |
5. |
Gonadotroph adenoma |
10-15% |
FSH-LH |
Inactive or hypogonadism |
6. |
Thyrotroph adenoma |
1% |
TSH |
Thyrotoxicosis |
7. |
Null cell adenoma/ oncocytoma |
20% |
Nil |
Pituitary failure |
8. |
Pleurihormonal adenoma |
15% |
Multiple hormones |
Mixed |
|
|
|
|
|
796
|
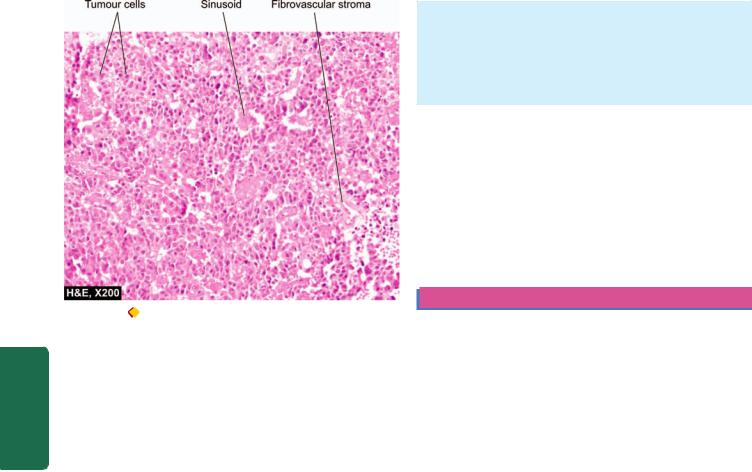
Figure 27.2 |
Pituitary adenoma, sinusoidal pattern. |
|
|
|
||
SECTION |
acidophil, basophil or chromophobe cells. These cells may |
||
have following 3 types of patterns: |
|||
|
|||
|
1. Diffuse pattern is composed of polygonal cells arranged |
||
|
in sheets with scanty stroma. |
||
|
2. Sinusoidal pattern consists of columnar or fusiform cells |
||
III |
with fibrovascular stroma around which the tumour cells |
||
are arranged (Fig. 27.2). |
|||
|
3. Papillary pattern is composed of columnar or fusiform |
||
Systemic |
cells arranged about fibrovascular papillae. |
||
Functionally, most common pituitary adenomas, in |
|||
|
|||
|
decreasing order of frequency, are: lactotroph (PRL-secreting) |
||
|
adenoma, somatotroph (GH-secreting) adenoma and |
||
Pathology |
corticotroph (ACTH-secreting) adenoma. Infrequently, |
||
mixed somatotroph-lactotroph (GH-PRL-secreting) |
|||
|
|||
|
adenoma, gonadotroph (FSH-LH-secreting) adenomas and |
||
|
null-cell (endocrinologically inactive) adenomas or |
||
|
oncocytoma are found. Pleurihormonal-pituitary adenoma, |
||
|
on the other hand, may have multiple hormone elaborations. |
||
|
Functional classification of pituitary adenoma can be done |
||
|
by carrying out specific immunostains against the hormone |
||
|
products. |
|
|
|
Pituitary adenoma may also occur as a part of multiple |
||
|
endocrine neoplasia type I (MEN-I) in which adenomas of |
||
|
pancreatic islets, parathyroids and the pituitary are found |
||
|
(page 829). Clinically, the patients are characterised by |
||
|
combination of features of Zollinger-Ellison’s syndrome, |
||
|
hyperparathyroidism and hyperpituitarism. |
||
|
Craniopharyngioma |
||
|
Craniopharyngioma is a benign tumour arising from |
||
|
remnants of Rathke’s pouch. It is more common in children |
||
|
and young adults. The tumour, though benign, compresses |
||
|
as well as invades the adjacent structures extensively. |
||
|
|
||
|
MORPHOLOGIC FEATURES. Grossly, the tumour is |
||
|
encapsulated, adherent to surrounding structures and is |
||
|
typically cystic, reddish-grey mass. |
||
|
|
|
|
Histologically, craniopharyngioma closely resembles ameloblastoma of the jaw (page 530). There are 2 distinct histologic features:
1.Stratified squamous epithelium frequently lining, a cyst and containing loose stellate cells in the centre; and
2.Solid ameloblastous areas.
Granular Cell Tumour (Choristoma)
Though tumours of the posterior pituitary are rare, granular cell tumour or choristoma is the most common tumour of the neurohypophysis. It is composed of a mass of cells having granular eosinophilic cytoplasm similar to the cells of the posterior pituitary. It arises as a result of developmental anomaly and hence the name choristoma. Generally, it remains asymptomatic and is discovered as an incidental autopsy finding.
ADRENAL GLAND
NORMAL STRUCTURE
ANATOMY. The adrenal glands lie at the upper pole of each kidney. Each gland weighs approximately 4 gm in the adult but in children the adrenals are proportionately larger. On sectioning, the adrenal is composed of 2 distinct parts: an outer yellow-brown cortex and an inner grey medulla. The anatomic and functional integrity of adrenal cortices are essential for life, while it does not hold true for adrenal medulla.
HISTOLOGY AND PHYSIOLOGY. Microscopically and functionally, cortex and medulla are quite distinct.
ADRENAL CORTEX. It is composed of 3 layers:
1.Zona glomerulosa is the outer layer and comprises about 10% of the cortex. It consists of cords or columns of polyhedral cells just under the capsule. This layer is responsible for the synthesis of mineralocorticoids, the most important of which is aldosterone, the salt and water regulating hormone.
2.Zona fasciculata is the middle layer and constitutes approximately 70% of the cortex. It is composed of columns of lipid-rich cells which are precursors of various steroid hormones manufactured in the adrenal cortex such as glucocorticoids (e.g. cortisol) and sex steroids (e.g. testosterone).
3.Zona reticularis is the inner layer which makes up the remainder of the adrenal cortex. It consists of cords of more compact cells than those of zona fasciculata but has similar functional characteristics of synthesis and secretion of
glucocorticoids and androgens.
The synthesis of glucocorticoids and adrenal androgens is under the control of ACTH from hypothalamus-anterior pituitary. In turn, ACTH release is under the control of a hypothalamic releasing factor called corticotropin-releasing factor. Release of aldosterone, on the other hand, is independent of ACTH control and is largely regulated by the serum levels of potassium and renin-angiotensin mechanism (page 98).

ADRENAL MEDULLA. The adrenal medulla is a component of the dispersed neuroendocrine system derived from primitive neuroectoderm; the other components of this system being paraganglia distributed in the vagi, paravertebral and visceral autonomic ganglia. The cells comprising this system are neuroendocrine cells, the major function of which is synthesis and secretion of catecholamines (epinephrine and norepinephrine). Various other peptides such as calcitonin, somatostatin and vasoactive intestinal polypeptide (VIP) are also secreted by these cells. The major metabolites of catecholamines are metanephrine, nor-me- tanephrine, vanillyl mandelic acid (VMA) and homovanillic acid (HVA). In case of damage to the adrenal medulla, its function is taken over by other paraganglia.
Diseases affecting the two parts of adrenal glands are quite distinctive in view of distinct morphology, and function of the adrenal cortex and medulla. While the disorders of the adrenal cortex include adrenocortical hyperfunction (hyperadrenalism), adrenocortical insufficiency (hypoadrenalism) and adrenocortical tumours, the main lesions affecting the adrenal medulla are the medullary tumours.
ADRENOCORTICAL HYPERFUNCTION (HYPERADRENALISM)
Hypersecretion of each of the three types of corticosteroids elaborated by the adrenal cortex causes distinct corresponding hyperadrenal clinical syndromes:
1.Cushing’s syndrome caused by excess of glucocorticoids (i.e. cortisol); also called chronic hypercortisolism.
2.Conn’s syndrome caused by oversecretion of mineralocorticoids (i.e. aldosterone); also called primary hyper-
aldosteronism.
3. Adrenogenital syndrome characterised by excessive production of adrenal sex steroids (i.e. androgens); also called adrenal
virilism.
Mixed forms of these clinical syndromes may also occur.
Cushing’s Syndrome (Chronic Hypercortisolism)
Cushing’s syndrome is caused by excessive production of cortisol of whatever cause. The full clinical expression of the syndrome, however, includes contribution of the secondary derangements.
ETIOPATHOGENESIS. There are 4 major etiologic types of Cushing’s syndrome which should be distinguished for effective treatment.
1. Pituitary Cushing’s syndrome. About 60-70% cases of Cushing’s syndrome are caused by excessive secretion of ACTH due to a lesion in the pituitary gland, most commonly a corticotroph adenoma or multiple corticotroph microadenomas. This group of cases was first described by Harvey Cushing, an American neurosurgeon, who termed the condition as Cushing’s disease. Also included in this group are cases with hypothalamic origin of excessive ACTH levels without apparent pituitary lesion. All cases with pituitary Cushing’s syndrome are characterised by bilateral adrenal cortical hyperplasia and elevated ACTH levels. These cases show therapeutic response on administration of high
doses of dexamethasone which suppresses ACTH secretion and causes fall in plasma cortisol level.
2.Adrenal Cushing’s syndrome. Approximately 20-25% cases of Cushing’s syndrome are caused by disease in one or both the adrenal glands. These include adrenal cortical adenoma, carcinoma, and less often, cortical hyperplasia. This group of cases is characterised by low serum ACTH levels and absence of therapeutic response to administration of high doses of glucocorticoid.
3.Ectopic Cushing’s syndrome. About 10-15% cases of Cushing’s syndrome have an origin in ectopic ACTH elaboration by non-endocrine tumours. Most often, the tumour is an oat cell carcinoma of the lung but other lung cancers, malignant thymoma and pancreatic tumours have also been implicated. The plasma ACTH level is high in these cases and cortisol secretion is not suppressed by dexamethasone administration.
4.Iatrogenic Cushing’s syndrome. Prolonged therapeutic administration of high doses of glucocorticoids or ACTH may result in Cushing’s syndrome e.g. in organ transplant recipients and in autoimmune diseases. These cases are generally associated with bilateral adrenocortical insufficiency.
CLINICAL FEATURES. Cushing’s syndrome occurs more often in patients between the age of 20-40 years with three times higher frequency in women than in men. The severity of the syndrome varies considerably, but in general the following features characterise a case of Cushing’s syndrome:
1.Central or truncal obesity contrasted with relatively thin arms and legs, buffalo hump due to prominence of fat over the shoulders, and rounded oedematous moon-face.
2.Increased protein breakdown resulting in wasting and thinning of the skeletal muscles, atrophy of the skin and subcutaneous tissue with formation of purple striae on the abdominal wall, osteoporosis and easy bruisability of the thin skin to minor trauma.
3.Systemic hypertension is present in 80% of cases because of associated retention of sodium and water.
4.Impaired glucose tolerance and diabetes mellitus are found in about 20% cases.
5.Amenorrhoea, hirsutism and infertility in many women.
6.Insomnia, depression, confusion and psychosis.
Conn’s Syndrome (Primary Hyperaldosteronism)
This is an uncommon syndrome occurring due to overproduction of aldosterone, the potent salt-retaining hormone.
ETIOPATHOGENESIS. The condition results primarily due to adrenocortical diseases as follows:
1.Adrenocortical adenoma, producing aldosterone.
2.Bilateral adrenal hyperplasia, especially in children (congenital hyperaldosteronism).
3.Rarely, adrenal carcinoma.
Primary hyperaldosteronism from any of the above causes is associated with low plasma renin levels. Secondary hyperaldosteronism, on the contrary, occurs in response to high plasma renin level due to overproduction of renin by the kidneys such as in renal ischaemia, reninoma or oedema.
797
System Endocrine The 27 CHAPTER

798CLINICAL FEATURES. Conn’s syndrome is more frequent in adult females. Its principal features are as under:
1.Hypertension, usually mild to moderate diastolic hypertension.
2.Hypokalaemia and associated muscular weakness, peripheral neuropathy and cardiac arrhythmias.
3.Retention of sodium and water.
4.Polyuria and polydipsia due to reduced concentrating power of the renal tubules.
Adrenogenital Syndrome (Adrenal Virilism)
Adrenal cortex secretes a smaller amount of sex steroids than the gonads. However, adrenocortical hyperfunction may occasionally cause sexual disturbances.
ETIOPATHOGENESIS. Hypersecretion of sex steroids, mainly androgens, may occur in children or in adults:
1.In children, it is due to congenital adrenal hyperplasia in which there is congenital deficiency of a specific enzyme.
2.In adults, it is caused by an adrenocortical adenoma or a carcinoma. Cushing’s syndrome is often present as well.
SECTION |
CLINICAL FEATURES. The clinical features depend upon |
|
the age and sex of the patient. |
||
1. In children, there is distortion of the external genitalia in |
||
girls, and precocious puberty in boys. |
||
2. In adults, the features in females show virilisation (e.g. |
||
hirsutism, oligomenorrhoea, deepening of voice, hyper- |
||
III |
trophy of the clitoris); and in males may rarely cause |
|
feminisation. |
||
|
||
Systemic |
3. There is generally increased excretion of 17-ketosteroids |
|
in the urine. |
||
ADRENOCORTICAL INSUFFICIENCY |
||
|
||
|
(HYPOADRENALISM) |
|
Pathology |
Adrenocortical insufficiency may result from deficient |
|
synthesis of cortical steroids from the adrenal cortex or may |
||
|
||
|
be secondary to ACTH deficiency. Three types of |
|
|
adrenocortical hypofunction are distinguished: |
|
|
1. Primary adrenocortical insufficiency caused primarily by the |
|
|
disease of the adrenal glands. Two forms are described: acute |
|
|
or ‘adrenal crisis’, and chronic or ‘Addison’s disease’. |
|
|
2. Secondary adrenocortical insufficiency resulting from |
|
|
diminished secretion of ACTH. |
|
|
3. Hypoaldosteronism characterised by deficient secretion of |
|
|
aldosterone. |
|
|
PRIMARY ADRENOCORTICAL INSUFFICIENCY |
|
|
Primary adrenal hypofunction occurs due to defect in the |
|
|
adrenal glands and normal pituitary function. It may develop |
|
|
in 2 ways: |
|
|
A. Acute primary adrenocortical insufficiency or ‘adrenal |
|
|
crisis’. |
|
|
B. Chronic primary adrenocortical insufficiency or |
|
|
‘Addison’s disease’. |
|
|
A. Primary Acute Adrenocortical Insufficiency |
|
|
(Adrenal Crisis) |
|
|
Sudden loss of adrenocortical function may result in an acute |
|
|
condition called adrenal crisis. |
ETIOPATHOGENESIS. Causes of acute insufficiency are as under:
1.Bilateral adrenalectomy e.g. in the treatment of cortical hyperfunction, hypertension and in selected cases of breast cancer.
2.Septicaemia e.g. in endotoxic shock and meningococcal infection producing grossly haemorrhagic and necrotic adrenal cortex termed adrenal apoplexy. The acute condition
so produced is called Waterhouse-Friderichsen’s syndrome.
3.Rapid withdrawal of steroids.
4.Any form of acute stress in a case of chronic insufficiency i.e. in Addison’s disease.
CLINICAL FEATURES. Clinical features of acute adrenocortical insufficiency are due to deficiency of mineralocorticoids and glucocorticoids. These are as follows:
1.Deficiency of mineralocorticoids (i.e. aldosterone deficiency) result in salt deficiency, hyperkalaemia and dehydration.
2.Deficiency of glucocorticoids (i.e. cortisol deficiency) leads to hypoglycaemia, increased insulin sensitivity and vomitings.
B.Primary Chronic Adrenocortical Insufficiency (Addison’s Disease)
Progressive chronic destruction of more than 90% of adrenal cortex on both sides results in an uncommon clinical condition called Addison’s disease.
ETIOPATHOGENESIS. Any condition which causes marked chronic adrenal destruction may produce Addison’s disease. These include: tuberculosis, autoimmune or idiopathic adrenalitis, histoplasmosis, amyloidosis, metastatic cancer, sarcoidosis and haemochromatosis. However, currently the first two causes—tuberculosis and autoimmune chronic destruction of adrenal glands, are implicated in majority of cases of Addison’s disease. Irrespective of the cause, the adrenal glands are bilaterally small and irregularly shrunken. Histologic changes, depending upon the cause, may reveal specific features as in tuberculosis and histoplasmosis, or the changes may be in the form of nonspecific lymphocytic infiltrate as in idiopathic (autoimmune) adrenalitis.
CLINICAL FEATURES. Clinical manifestations develop slowly and insidiously. The usual features are as under:
1.Asthenia i.e. progressive weakness, weight loss and lethargy as the cardinal symptoms.
2.Hyperpigmentation, initially most marked on exposed areas, but later involves unexposed parts and mucous membranes as well.
3.Arterial hypotension.
4.Vague upper gastrointestinal symptoms such as mild loss of appetite, nausea, vomiting and upper abdominal pain.
5.Lack of androgen causing loss of hair in women.
6.Episodes of hypoglycaemia.
7.Biochemical changes include reduced GFR, acidosis, hyperkalaemia and low levels of serum sodium, chloride and bicarbonate.

SECONDARY ADRENOCORTICAL INSUFFICIENCY
Adrenocortical insufficiency resulting from deficiency of ACTH is called secondary adrenocortical insufficiency.
ETIOPATHOGENESIS. ACTH deficiency may appear in 2 settings :
1.Selective ACTH deficiency due to prolonged administration of high doses of glucocorticoids. This leads to suppression of ACTH release from the pituitary gland and selective deficiency.
2.Panhypopituitarism due to hypothalamus-pituitary diseases is associated with deficiency of multiple trophic hormones (page 794).
CLINICAL FEATURES. The clinical features of secondary adrenocortical insufficiency are like those of Addison’s disease except the following:
1.These cases lack hyperpigmentation because of suppressed production of melanocyte-stimulating hormone (MSH) from the pituitary.
2.Plasma ACTH levels are low-to-absent in secondary insufficiency but are elevated in Addison’s disease.
3.Aldosterone levels are normal due to stimulation by renin.
HYPOALDOSTERONISM
Isolated deficiency of aldosterone with normal cortisol level may occur in association with reduced renin secretion.
ETIOPATHOGENESIS. The causes of such hyporeninism are as follows:
1.Congenital defect due to deficiency of an enzyme required for its synthesis.
2.Prolonged administration of heparin.
3.Certain diseases of the brain.
4.Excision of an aldosterone-secreting tumour.
CLINICAL FEATURES. The patients of isolated hypoaldosteronism are adults with mild renal failure and diabetes mellitus. The predominant features are hyperkalaemia and metabolic acidosis.
TUMOURS OF ADRENAL GLANDS
Primary tumours of the adrenal glands are uncommon and include distinct adrenocortical tumours and medullary tumours. However, adrenal gland is a more common site for metastatic carcinoma.
ADRENOCORTICAL TUMOURS
Cortical Adenoma
The commonest cortical tumour is adenoma. They are indistinguishable from hyperplastic nodules except that lesions smaller than 2 cm diameter are labelled hyperplastic nodules. A cortical adenoma is a benign and slow-growing tumour. It is usually small and nonfunctional. A few large adenomas may, however, produce excess of cortisol, aldosterone or androgen. Association of cortical adenomas with systemic hypertension has been suggested by some
workers. Occasionally, a cortical adenoma may be a part of multiple endocrine neoplasia type I (MEN-I) in which patients have associated adenomas of parathyroid, islet cells and anterior pituitary (page 829).
MORPHOLOGIC FEATURES. Grossly, an adenoma is usually a small, solitary, spherical and encapsulated tumour which is well-delineated from the surrounding normal adrenal gland. Cut section is typically bright yellow.
Microscopically, the tumour cells are arranged in trabeculae and generally resemble the cells of zona fasciculata. Less frequently, the cells of adenoma are like those of zona glomerulosa or zona reticularis.
Cortical Carcinoma
Carcinoma of the adrenal cortex is an uncommon tumour occurring mostly in adults. It invades locally as well as spreads to distant sites. Most cortical carcinomas secrete one of the adrenocortical hormones excessively.
MORPHOLOGIC FEATURES. Grossly, an adrenal carcinoma is generally large, spherical and welldemarcated tumour. On cut section, it is predominantly yellow with intermixed areas of haemorrhages, necrosis and calcification.
Microscopically, the cortical carcinoma may vary from well-differentiated to anaplastic growth. Welldifferentiated carcinoma consists of foci of atypia in an adenoma, while anaplastic carcinoma shows large, pleomorphic and bizarre cells with high mitotic activity.
MEDULLARY TUMOURS
The most significant lesions of the adrenal medulla are neoplasms. These include the following:

 Benign tumours: These are less common and include pheochromocytoma and myelolipoma.
Benign tumours: These are less common and include pheochromocytoma and myelolipoma.
 Tumours arising from embryonic nerve cells: These are more common and include neuroblastoma and ganglioneuroma.
Tumours arising from embryonic nerve cells: These are more common and include neuroblastoma and ganglioneuroma.
These tumours together with extra-adrenal paraganglioma are described below.
Pheochromocytoma (Chromaffin Tumour)
Pheochromocytoma (meaning dusky brown tumour) is generally a benign tumour arising from the pheochromocytes (i.e. chromaffin cells) of the adrenal medulla. The extraadrenal pheochromocytomas arising from other paraganglia are preferably called paragangliomas, named along with the anatomic site of origin, as described later.
Pheochromocytoma may occur at any age but most patients are 20-60 years old. Most pheochromocytomas are slow-growing and benign but about 5% of the tumours are malignant, invasive and metastasising. These tumours are commonly sporadic but 10-20% are associated with familial syndromes of multiple endocrine neoplasia (MEN) having bilaterality and association with medullary carcinoma of the thyroid, hyperparathyroidism, pituitary adenoma, mucosal
799
System Endocrine The 27 CHAPTER
800neuromas and von Recklinghausen’s neurofibromatosis in varying combinations.
The clinical features of pheochromocytoma are predominantly due to secretion of catecholamines, both epinephrine and norepinephrine. The most common feature is hypertension. Other manifestations due to sudden release of catecholamines are congestive heart failure, myocardial infarction, pulmonary oedema, cerebral haemorrhage, and even death. The diagnosis is established by measuring 24hour urinary catecholamines or their metabolites such as metanephrine and VMA.
|
MORPHOLOGIC FEATURES. Grossly, the tumour is |
|
|
soft, spherical, may be quite variable in size and weight, |
|
|
and well-demarcated from the adjacent adrenal gland. On |
|
|
cut section, the tumour is grey to dusky brown with areas |
|
|
of haemorrhages, necrosis, calcification and cystic change |
|
|
(Fig. 27.3). On immersing the tumour in dichromate |
|
|
fixative, it turns brown-black due to oxidation of |
|
|
catecholamines in the tumour and hence the name |
|
|
chromaffin tumour. |
|
|
Microscopically, the tumour has the following |
|
SECTION |
characteristics (Fig. 27.4): |
|
1. The tumour cells are arranged characteristically as |
||
|
||
|
well-defined nests (also termed as zellballen pattern) |
|
|
separated by abundant fibrovascular stroma. |
|
|
2. Other arrangements are as solid columns, sheets, |
|
III |
trabeculae or clumps. |
|
3. The tumour cells are large, polyhedral and |
||
|
pleomorphic with abundant granular amphophilic or |
|
PathologySystemic |
basophilic cytoplasm and vesicular nuclei. |
|
4. The tumour cells of pheochromocytoma stain |
||
|
||
|
positively with neuroendocrine substances such as |
|
|
neuron-specific enolase (NSE) and chromogranin. |
|
|
|
About 10% of pheochromcytomas may be malignant having tendency for osseous metastases.
Myelolipoma
Myelolipoma is an uncommon benign adrenal medullary tumour found incidentally at autopsy. Less often, it may produce symptoms due to excessive hormone elaboration.
MORPHOLOGIC FEATURES. Grossly, a myelolipoma is usually a small tumour, measuring 0.2-2 cm in diameter. Microscopically, it consists of well-differentiated adipose tissue in which is scattered clumps of haematopoietic cells are seen.
Neuroblastoma
Neuroblastoma, also called as sympathicoblastoma, is a common malignant tumour of embryonic nerve cells, occurring most commonly in children under 5 years of age. Vast majority of cases occur within the abdomen (in the adrenal medulla and paravertebral autonomic ganglia) and rarely in the cerebral hemisphere. Most cases are sporadic but familial occurrence with autosomal dominant transmission is also seen.
The clinical manifestations of neuroblastoma are related to its rapid local growth, metastatic spread or development of hormonal syndrome. Local symptoms include abdominal distension, fever, weight loss and malaise. Foci of calcification may be observed on radiologic examination of the abdomen. Metastatic spread occurs early and widely through haematogenous as well as lymphatic routes and involves bones (especially skull), liver, lungs and regional lymph nodes. Neuroblastoma produces variable amounts of catecholamines and its metabolites such as vanillyl mandelic acid (VMA) and homovanillic acid (HVA), which can be detected in the 24-hour urine. Less often, the patient develops carcinoid-like syndrome, probably due to production of kinins or prostaglandins by the tumour. The features in such a case include watery diarrhoea, flushing of the skin and
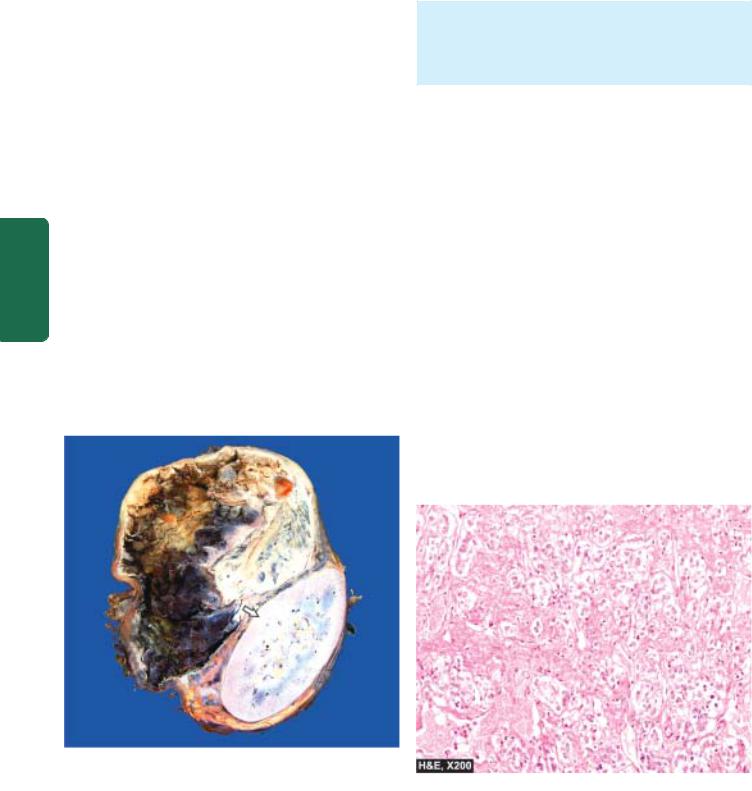
Figure 27.3 
 Pheochromocytoma of the adrenal medulla. The specimen shows compressed kidney at the lower end (arrow) while the upper end shows a large spherical tumour separate from the kidney. Cut surface of tumour shows cystic change while solid areas show dark brown, necrotic and haemorrhagic tumour.
Pheochromocytoma of the adrenal medulla. The specimen shows compressed kidney at the lower end (arrow) while the upper end shows a large spherical tumour separate from the kidney. Cut surface of tumour shows cystic change while solid areas show dark brown, necrotic and haemorrhagic tumour.
Figure 27.4 
 Adrenal pheochromocytoma. The tumour has typical zellballen or nested pattern. The tumour cells are large, polyhedral and pleomorphic having abundant granular cytoplasm.
Adrenal pheochromocytoma. The tumour has typical zellballen or nested pattern. The tumour cells are large, polyhedral and pleomorphic having abundant granular cytoplasm.
801
Figure 27.5 |
Neuroblastoma, It shows small, round to oval cells forming irregular sheets separated by fibrovascular stroma. A few Homer- |
Wright’s pseudorosettes are also present. Inset shows a close-up view of pseudorosette.
hypokalaemia. Rarely, the tumour may produce sufficient catecholamines to cause hypertension.
MORPHOLOGIC FEATURES. Grossly, the tumour is generally large, soft and lobulated mass with extensive areas of necrosis and haemorrhages. The tumour is usually diffusely infiltrating into the adjacent tissues. Cut surface of the tumour is grey white and may reveal minute foci of calcification.
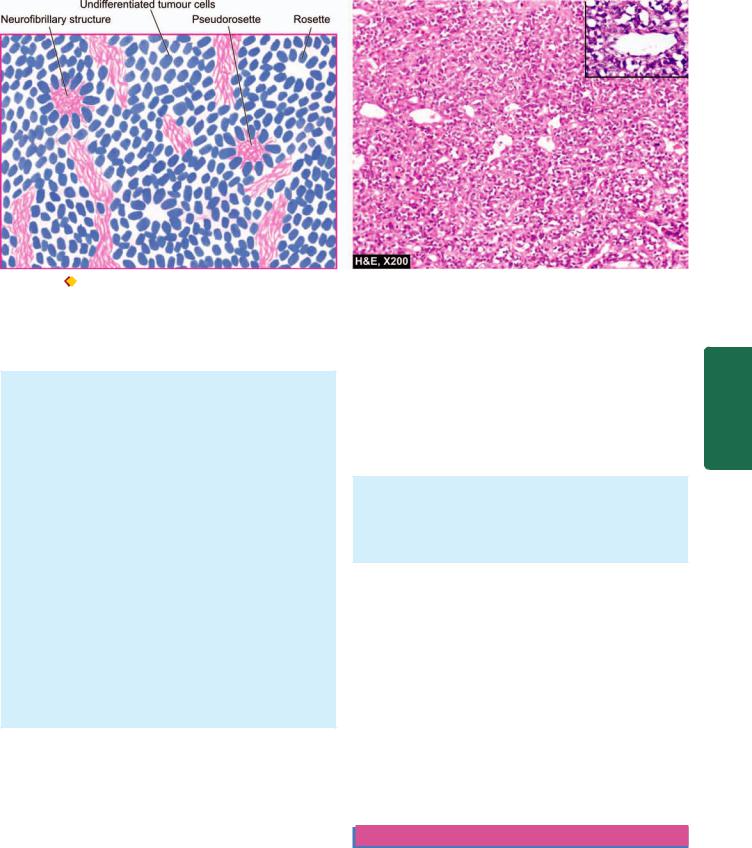
Microscopically, neuroblastoma has the following characteristics (Fig. 27.5):
1.The tumour cells are small, round and oval, slightly larger than lymphocytes, and have scanty and poorlydefined cytoplasm and hyperchromatic nuclei.
2.They are generally arranged in irregular sheets separated by fibrovascular stroma.
3.Classical neuroblastomas show Homer-Wright’s rosettes (pseudorosettes) which have a central fibrillar eosinophilic material surrounded by radially arranged tumour cells. The central fibrillar material stains positively by silver impregnation methods indicating their nature as young nerve fibrils.
4.The tumour cells stain positively with immunohistochemical markers such as neuron-specific enolase (NSE), neurofilaments (NF) and chromogranin.
Prognosis of neuroblastoma depends upon a few variables:
i)Age of the child below 2 years is associated with better prognosis.
ii)Extra-abdominal location of the tumour have better outlook than abdominal masses.
iii)Patients in clinical stage I (confined to the organ of origin) or stage II (tumour extending in continuity beyond the organ of origin but not crossing the midline) have better prognosis than higher stages with distant metastases.
iv)Tumours with amplification of MYC oncogene and p53 are associated with poor prognosis.
Ganglioneuroma
A ganglioneuroma is a mature, benign and uncommon tumour occurring in adults. It is derived from ganglion cells, most often in the posterior mediastinum, and uncommonly in other peripheral ganglia and brain. The tumour produces symptoms because of its size and location. Catecholamines and their metabolites can be detected in large amounts in the 24-hour urine specimen of patients with ganglioneuroma.
MORPHOLOGIC FEATURES. Grossly, the tumour is spherical, firm and encapsulated.
Microscopically, it contains large number of well-formed ganglionic nerve cells scattered in fibrillar stroma and myelinated and non-myelinated nerve fibres.
Extra-adrenal Paraganglioma (Chemodectoma)
Parasympathetic paraganglia located in extra-adrenal sites such as the carotid bodies, vagus, jugulotympanic and aorticosympathetic (pre-aortic) paraganglia may produce neoplasms, collectively termed paragangliomas with the anatomic site of origin e.g. carotid body paraganglioma, intravagal paraganglioma, jugulotympanic paraganglioma etc. These tumours are also called chemodectomas because of their responsiveness to chemoreceptors. They are uncommon tumours found in adults and rarely secrete excess of catecholamines, except aorticosympathetic paraganglioma (also termed extra-adrenal pheochromocytoma). Paragangliomas are generally benign but recurrent tumours. A small proportion of them may metastasise widely.
THYROID GLAND
NORMAL STRUCTURE
ANATOMY. Embryologically, the thyroid gland arises from a midline invagination at the root of the tongue and grows downwards in front of trachea and thyroid cartilage to reach its normal position. Failure to descent may produce
System Endocrine The 27 CHAPTER

802 anomalous lingual thyroid. The thyroglossal duct that connects the gland to the pharyngeal floor normally disappears by 6th week of embryonic life. In adults, its proximal end is represented by foramen caecum at the base of the tongue and distal end by the pyramidal lobe of the thyroid. Persistence of the remnants of thyroglossal duct in the adults may develop into thyroglossal cyst (page 520). The C-cells of the thyroid originate from the neuroectoderm.
The thyroid gland in an adult weighs 15-40 gm and is composed of two lateral lobes connected in the midline by a broad isthmus which may have a pyramidal lobe extending upwards. Cut section of normal thyroid is yellowish and translucent.
|
HISTOLOGY. The thyroid is composed of lobules of colloid- |
|
|
filled spherical follicles or acini. The lobules are enclosed by |
|
|
fibrovascular septa. The follicles are the main functional units |
|
|
of the thyroid. They are lined by cuboidal epithelium with |
|
|
numerous fine microvilli extending into the follicular colloid |
|
|
that contains the glycoprotein, thyroglobulin. The follicles are |
|
|
separated from each other by delicate fibrous tissue that |
|
|
contains blood vessels, lymphatics and nerves. Calcitonin- |
|
SECTION |
secreting C-cells or parafollicular cells are dispersed within |
|
the follicles and can only be identified by silver stains and |
||
of iodine-containing thyroid hormones, thyroxine (T4) and |
||
|
immunohistochemical methods. |
|
|
FUNCTIONS. The major function of the thyroid gland is to |
|
|
maintain a high rate of metabolism which is done by means |
|
III |
tri-iodothyronine (T3). |
|
|
The thyroid is one of the most labile organs in the body |
|
Systemic |
and responds to numerous stimuli such as puberty, |
|
pregnancy, physiologic stress and various pathologic states. |
||
|
||
|
This functional lability of the thyroid is responsible for |
|
|
transient hyperplasia of the thyroidal epithelium. Under |
|
|
normal conditions, the epithelial lining of the follicles may |
|
Pathology |
show changes in various phases of function as under: |
|
thyroidism. |
||
|
1. Resting phase is characterised by large follicles lined by |
|
|
flattened cells and filled with deeply staining homogeneous |
|
|
colloid e.g. in colloid goitre and iodine-treated hyper- |
|
|
2. Secretory phase in which the follicles are lined by |
|
|
cuboidal epithelium and the colloid is moderately dark pink |
|
|
e.g. in normal thyroid. |
|
|
3. Resorptive phase is characterised by follicles lined by |
|
|
columnar epithelium and containing lightly stained |
|
|
vacuolated and scalloped colloid e.g. in hyperthyroidism. |
|
|
The synthesis and release of the two main circulating |
|
|
thyroid hormones, T3 and T4 are regulated by hypophyseal |
|
|
thyroid-stimulating hormone (TSH) and involves the |
|
|
following steps: |
|
|
1. Iodine trapping by thyroidal cells involves absorbing of |
|
|
iodine from the blood and concentrating it more than twenty- |
|
|
fold. |
|
|
2. Oxidation of the iodide takes place within the cells by a |
|
|
thyroid peroxidase. |
|
|
3. Iodination occurs next, at the microvilli level between |
|
|
the oxidised iodine and the tyrosine residues of thyroglobulin |
|
|
so as to form mono-iodotyrosine (MIT) and di-iodotyrosine |
|
|
(DIT). |
4. Coupling of MIT and DIT in the presence of thyroid peroxidase forms tri-iodothyronine (T3) and thyroxine (T4).
The thyroid hormones so formed are released by endocytosis of colloid and proteolysis of thyroglobulin within the follicular cells resulting in discharge of T3 and T4 into circulation where they are bound to thyroxine-binding globulin.
A number of thyroid function tests are currently available. These include the following:

 Determination of serum levels of T3, T4 by radioimmunoassay (RIA).
Determination of serum levels of T3, T4 by radioimmunoassay (RIA).

 TSH and TRH determination.
TSH and TRH determination.

 Determination of calcitonin secreted by parafollicular C cells.
Determination of calcitonin secreted by parafollicular C cells.
 Estimation of thyroglobulin secreted by thyroid follicular cells.
Estimation of thyroglobulin secreted by thyroid follicular cells.

 Assessment of thyroid activity by its ability to uptake radioactive iodine (RAIU).
Assessment of thyroid activity by its ability to uptake radioactive iodine (RAIU).
 Assessment whether thyroid lesion is a nonfunctioning (‘cold nodule’) or hyperactive mass (‘hot nodule’).
Assessment whether thyroid lesion is a nonfunctioning (‘cold nodule’) or hyperactive mass (‘hot nodule’).
Diseases of the thyroid include: functional disorders (hyperthyroidism and hypothyroidism), thyroiditis, Graves’ disease, goitre and tumours. The relative frequency of some of these diseases varies in different geographic regions according to the iodine content of the diet consumed. One of the important investigation tools available in current times is the widespread use of FNAC for thyroid lesions which helps in avoiding a large number of unwanted diagnostic biopsies.
FUNCTIONAL DISORDERS
Two significant functional disorders characterised by distinct clinical syndromes are described. These are: hyperthyroidism
(thyrotoxicosis) and hypothyroidism.
HYPERTHYROIDISM (THYROTOXICOSIS)
Hyperthyroidism, also called thyrotoxicosis, is a hypermetabolic clinical and biochemical state caused by excess production of thyroid hormones. The condition is more frequent in females and is associated with rise in both T3 and T4 levels in blood, though the increase in T3 is generally greater than that of T4.
ETIOPATHOGENESIS. Hyperthyroidism may be caused by many diseases but three most common causes are: Graves’ disease (diffuse toxic goitre), toxic multinodular goitre and a toxic adenoma. Less frequent causes are hypersecretion of pituitary TSH by a pituitary tumour, hypersecretion of TRH, thyroiditis, metastatic tumours of the thyroid, struma ovarii, congenital hyperthyroidism in the newborn of mother with Graves’ disease, hCG-secreting tumours due to mild thyrotropic effects of hCG (e.g. hydatidiform mole, choriocarcinoma and testicular tumours), and lastly, by excessive doses of thyroid hormones or iodine called jodbasedow disease.
CLINICAL FEATURES. Patients with hyperthyroidism have a slow and insidious onset, varying in severity from case to case. The usual symptoms are emotional instability,
nervousness, palpitation, fatigue, weight loss in spite of good appetite, heat intolerance, perspiration, menstrual disturbances and fine tremors of the outstretched hands. Cardiac manifestations in the form of tachycardia, palpitations and cardiomegaly are invariably present in hyperthyroidism. The skin of these patients is warm, moist and flushed. Weakness of skeletal muscles and osteoporosis are common. Typical eye changes in the form of exophthalmos are a common feature in Graves’ disease. Serum levels of T3 and T4 are elevated but TSH secretion is usually inhibited.
A sudden spurt in the severity of hyperthyroidism termed
‘thyroid storm’ or ‘thyroid crisis’ may occur in patients who have undergone subtotal thyroidectomy before adequate control of hyperthyroid state, or in a hyperthyroid patient under acute stress, trauma, and with severe infection. These patients develop high grade fever, tachycardia, cardiac arrhythmias and coma and may die of congestive heart failure or hyperpyrexia.
HYPOTHYROIDISM
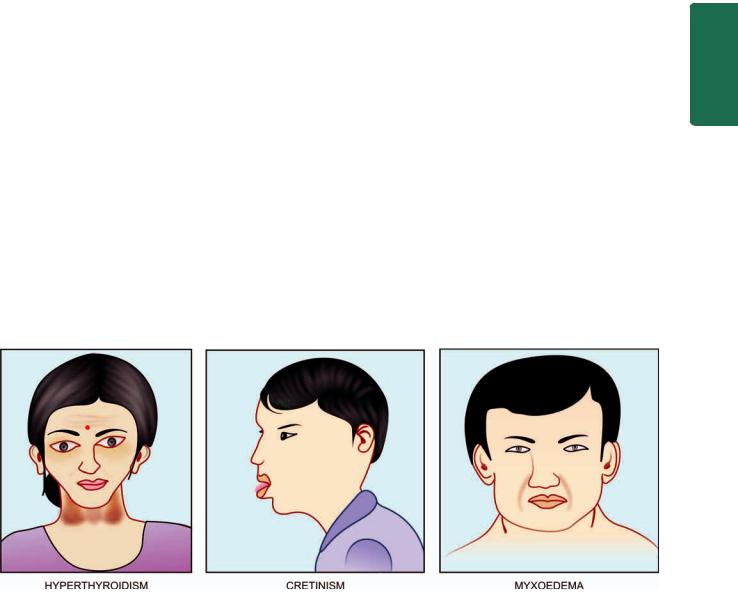
Hypothyroidism is a hypometabolic clinical state resulting from inadequate production of thyroid hormones for prolonged periods, or rarely, from resistance of the peripheral tissues to the effects of thyroid hormones. The clinical manifestations of hypothyroidism, depending upon the age at onset of disorder, are divided into 2 forms:
1.Cretinism or congenital hypothyroidism is the development of severe hypothyroidism during infancy and childhood.
2.Myxoedema is the adulthood hypothyroidism.
Cretinism
A cretin is a child with severe hypothyroidism present at birth or developing within first two years of postnatal life. This is the period when brain development is taking place; in the absence of treatment the child is both physically and mentally retarded. The word ‘Cretin’ is derived from the French, meaning Christ-like because these children are so mentally retarded that they are incapable of committing sins.
ETIOPATHOGENESIS. The causes of congenital hypothyroidism are as follows:
1.Developmental anomalies e.g. thyroid agenesis and ectopic thyroid.
2.Genetic defect in thyroid hormone synthesis e.g. defect in iodine trapping, oxidation, iodination, coupling and thyroglobulin synthesis.
3.Foetal exposure to iodides and antithyroid drugs.
4.Endemic cretinism in regions with endemic goitre due to dietary lack of iodine (sporadic cretinism, on the other hand, is due to developmental anomalies and genetic defects in thyroid hormone synthesis described above).
CLINICAL FEATURES. The clinical manifestations usually become evident within a few weeks to months of birth. The presenting features of a cretin are: slow to thrive, poor feeding, constipation, dry scaly skin, hoarse cry and bradycardia. As the child ages, clinical picture of fullydeveloped cretinism emerges characterised by impaired skeletal growth and consequent dwarfism, round face, narrow forehead, widely-set eyes, flat and broad nose, big protuberant tongue and protuberant abdomen. Neurological features such as deaf-mutism, spasticity and mental deficiency are more evident in sporadic cretinism due to developmental anomalies and dyshormonogenetic defects.
Characteristic laboratory findings include a rise in TSH level and fall in T3 and T4 levels.
Myxoedema
The adult-onset severe hypothyroidism causes myxoedema. The term myxoedema connotes non-pitting oedema due to accumulation of hydrophilic mucopolysaccharides in the ground substance of dermis and other tissues.
ETIOPATHOGENESIS. There are several causes of myxoedema listed below but the first two are the most common causes:
1.Ablation of the thyroid by surgery or radiation.
2.Autoimmune (lymphocytic) thyroiditis (termed primary idiopathic myxoedema).
3.Endemic or sporadic goitre.
4.Hypothalamic-pituitary lesions.
5.Thyroid cancer.
6.Prolonged administration of antithyroid drugs.
7.Mild developmental anomalies and dyshormonogenesis.
803
System Endocrine The 27 CHAPTER
Figure 27.6 
 Appearance in functional disorders of the thyroid gland.
Appearance in functional disorders of the thyroid gland.
804CLINICAL FEATURES. The onset of myxoedema is slow and a fully-developed clinical syndrome may appear after several years of hypothyroidism. The striking features are cold intolerance, mental and physical lethargy, constipation, slowing of speech and intellectual function, puffiness of face, loss of hair and altered texture of the skin.
The laboratory diagnosis in myxoedema is made by low
serum T3 and T4 levels and markedly elevated TSH levels as in the case of cretinism but cases with suprathyroid lesions (hypothalamic-pituitary disease) have low TSH levels.
The clinical appearance of these three major forms of functional disorders of the thyroid gland is shown in
Fig. 27.6.
THYROIDITIS
Inflammation of the thyroid, thyroiditis, is more often due to non-infectious causes and is classified on the basis of onset and duration of disease into acute, subacute and chronic as under:
|
I. |
Acute thyroiditis: |
|
1. |
Bacterial infection e.g. Staphylococcus, Streptococcus. |
SECTION |
2. Fungal infection e.g. Aspergillus, Histoplasma, Pneumocystis. |
|
thyroiditis, giant cell thyroiditis, viral thyroiditis) |
||
|
3. |
Radiation injury |
II. Subacute thyroiditis:
1. Subacute granulomatous thyroiditis (de Quervain’s
III |
2. |
Subacute lymphocytic (postpartum, silent) thyroiditis |
|
3. |
Tuberculous thyroiditis |
||
|
|||
Systemic |
III. Chronic thyroiditis: |
||
1. |
Autoimmune thyroiditis (Hashimoto’s thyroiditis or |
||
chronic lymphocytic thyroiditis) |
|||
2. Riedel’s thyroiditis (or invasive fibrous thyroiditis). |
|||
|
While acute infectious thyroiditis is uncommon, some of |
||
Pathology |
|
||
the morphologically important forms of thyroiditis from the |
|||
above list are discussed below. |
|||
HASHIMOTO’S (AUTOIMMUNE, |
|||
CHRONIC LYMPHOCYTIC) THYROIDITIS |
|||
|
|||
Hashimoto’s thyroiditis, also called diffuse lymphocytic thyroiditis, struma lymphomatosa or goitrous autoimmune thyroiditis, is characterised by 3 principal features:
1.Diffuse goitrous enlargement of the thyroid.
2.Lymphocytic infiltration of the thyroid gland.
3.Occurrence of thyroid autoantibodies.
Hashimoto’s thyroiditis occurs more frequently between the age of 30 and 50 years and shows an approximately tenfold preponderance among females. Though rare in children, about half the cases of adolescent goitre are owing to autoimmune thyroiditis. Hashimoto’s thyroiditis is the most common cause of goitrous hypothyroidism in regions where iodine supplies are adequate. Regions where iodine intake is highest have higher incidence of Hashimoto’s thyroiditis e.g. in Japan and the United States.
ETIOPATHOGENESIS. Hashimoto’s thyroiditis is an autoimmune disease is well established. Hashimoto, a Japanese surgeon, described it in 1912 as the first auto-
immune disease of any organ. Autoimmune pathogenesis of Hashimoto’s thyroiditis is explained by the following observations:
1.Other autoimmune disease association: Like in other autoimmune diseases, Hashimoto’s disease has been found in association with other autoimmune diseases such as Graves’ disease, SLE, Sjögren’s syndrome, rheumatoid arthritis, pernicious anaemia and Type 1 diabetes mellitus.
2.Immune destruction of thyroid cells: The sequence of immune phenomena is initial activation of CD4+ T helper cells. These cells then induce infiltration of CD8+ T cytotoxic cells in the thyroid parenchyma as well as activate B cells to form autoantibodies, which bring about immune destruction of thyroid parenchyma.
3.Detection of autoantibodies: The following autoantibodies against different thyroid cell antigens are detectable in the sera of most patients with Hashimoto’s thyroiditis:
i)Thyroid microsomal autoantibodies (against the microsomes of the follicular cells).
ii)Thyroglobulin autoantibodies.
iii)TSH receptor autoantibodies.
iv)Less constantly found are thyroid autoantibodies against follicular cell membranes, thyroid hormones themselves, and colloid component other than thyroglobulin.
4.Inhibitory TSH-receptor antibodies: TSH-receptor antibody seen on the surface of thyroid cells in Hashimoto’s thyroiditis is inhibitory to TSH, producing hypothyroidism. Similar antibody is observed in Graves’ disease where it causes hyperthyroidism. It appears that TSH-receptor antibody may act both to depress or stimulate the thyroid cells to produce hypoor hyperthyroidism respectively. Thus, these patients may have alternate episodes of hypoor hyperthyroidism.
5.Genetic basis: The disease has higher incidence in firstdegree relatives of affected patients. Hashimoto’s thyroiditis is seen more often with HLA-DR3 and HLA-DR5 subtypes.
MORPHOLOGIC FEATURES. Pathologically, two varieties of Hashimoto’s thyroiditis are seen: classic form, the usual and more common, and fibrosing variant found in only 10% cases of Hashimoto’s thyroiditis.
Grossly, the classic form is characterised by diffuse, symmetric, firm and rubbery enlargement of the thyroid which may weigh 100-300 gm. Sectioned surface of the thyroid is fleshly with accentuation of normal lobulations but with retained normal shape of the gland. The fibrosing variant has a firm, enlarged thyroid with compression of the surrounding tissues.
Histologically, the classic form shows the following features (Fig. 27.7):
1.There is extensive infiltration of the gland by lymphocytes, plasma cells, immunoblasts and macrophages, with formation of lymphoid follicles having germinal centres.
2.There is decreased number of thyroid follicles which are generally atrophic and are often devoid of colloid.
3.The follicular epithelial cells are transformed into their degenerated state termed Hurthle cells (also called
805
Figure 27.7 
 Hashimoto’s thyroiditis. Histologic features include: lymphoid cell infiltration with formation of lymphoid follicles having germinal centres; small, atrophic and colloid-deficient follicles; presence of Hurthle cells which have granular oxyphil cytoplasm and large irregular nuclei; and slight fibrous thickening of lobular septa.
Hashimoto’s thyroiditis. Histologic features include: lymphoid cell infiltration with formation of lymphoid follicles having germinal centres; small, atrophic and colloid-deficient follicles; presence of Hurthle cells which have granular oxyphil cytoplasm and large irregular nuclei; and slight fibrous thickening of lobular septa.
Askanazy cells, or oxyphil cells, or oncocytes). These cells have abundant oxyphilic or eosinophilic and granular cytoplasm due to large number of mitochondria and contain large bizarre nuclei.
4. There is slight fibrous thickening of the septa separating the thyroid lobules.
The less common fibrosing variant of Hashimoto’s thyroiditis shows considerable fibrous replacement of thyroid parenchyma and a less prominent lymphoid infiltrate.
CLINICAL FEATURES. The presenting feature of Hashimoto’s thyroiditis is a painless, firm and moderate goitrous enlargement of the thyroid gland, usually associated with hypothyroidism, in an elderly woman. At this stage, serum T3 and T4 levels are decreased and RAIU is also reduced. A few cases, however, develop hyperthyroidism, termed hashitoxicosis, further substantiating the similarities in the autoimmune phenomena between Hashimoto’s thyroiditis and Graves’ thyrotoxicosis. There is no increased risk of developing thyroid carcinoma in Hashimoto’s thyroiditis but there is increased frequency of malignant lymphoma in these cases.
SUBACUTE LYMPHOCYTIC THYROIDITIS
Subacute lymphocytic (or painless or silent or postpartum) thyroiditis is another variety of autoimmune thyrioditis. Clinically, it differs from subacute granulomatous thyroiditis in being non-tender thyroid enlargement. It is seen more often 3-6 months after delivery.
Microscopically, the features are as under:
1.Dense multifocal infiltrate of lymphocytes and plasma cells in the parenchyma.
2.Collapse of thyroid follicles.
3.Rarely, presence of lymphoid follicles with germinal centres, simulating Hashimoto’s thyroiditis.
SUBACUTE GRANULOMATOUS (DE QUERVAIN’S) THYROIDITIS
Granulomatous thyroiditis, also called de Quervain’s or subacute, or giant cell thyroiditis, is a distinctive form of selflimited inflammation of the thyroid gland. Etiology of the condition is not known but clinical features of a prodromal phase and preceding respiratory infection suggest a possible viral etiology. The disease is more common in young and middle-aged women and may present clinically with painful moderate thyroid enlargement with fever, features of hyperthyroidism in the early phase of the disease, and hypothyroidism if the damage to the thyroid gland is extensive. The condition is self-limiting and shows complete recovery of thyroid function in about 6 months.
MORPHOLOGIC FEATURES. Grossly, there is moderate enlargement of the gland which is often asymmetric or focal. The cut surface of the involved area is firm and yellowish-white.
Microscopically, the features vary according to the stage of the disease:
 Initially, there is acute inflammatory destruction of the thyroid parenchyma and formation of microabscesses.
Initially, there is acute inflammatory destruction of the thyroid parenchyma and formation of microabscesses.  Later, the more characteristic feature of granulomatous appearance is produced. These granulomas consist of central colloid material surrounded by histiocytes and scattered multinucleate giant cells.
Later, the more characteristic feature of granulomatous appearance is produced. These granulomas consist of central colloid material surrounded by histiocytes and scattered multinucleate giant cells.
 More advanced cases may show fibroblastic proliferation.
More advanced cases may show fibroblastic proliferation.
Morphologically similar appearance may be produced in cases where vigorous thyroid palpation may initiate mechanical trauma to follicles, so-called palpation thyroiditis.
RIEDEL’S THYROIDITIS
Riedel’s thyroiditis, also called Riedel’s struma or invasive fibrous thyroiditis, is a rare chronic disease characterised by
System Endocrine The 27 CHAPTER
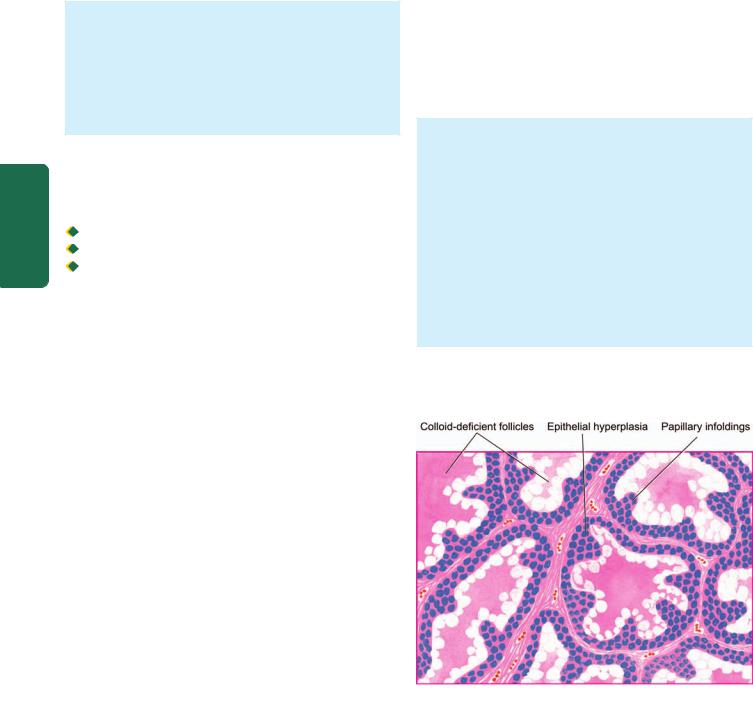
806stony-hard thyroid that is densely adherent to the adjacent structures in the neck. The condition is clinically significant due to compressive clinical features (e.g. dysphagia, dyspnoea, recurrent laryngeal nerve paralysis and stridor) and resemblance with thyroid cancer. Riedel’s struma is seen more commonly in females in 4th to 7th decades of life. The etiology is unknown but possibly Riedel’s thyroiditis is a part of multifocal idiopathic fibrosclerosis (page 591). This group of disorders includes: idiopathic retroperitoneal, mediastinal and retro-orbital fibrosis, and sclerosing cholangitis, all of which may occur simultaneously with Riedel’s thyroiditis.
MORPHOLOGIC FEATURES. Grossly, the thyroid gland is usually contracted, stony-hard, asymmetric and firmly adherent to the adjacent structures. Cut section is hard and devoid of lobulations.
Microscopically, there is extensive fibrocollagenous replacement, marked atrophy of the thyroid parenchyma, focally scattered lymphocytic infiltration and invasion of the adjacent muscle tissue by the process.
|
GRAVES’ DISEASE (DIFFUSE TOXIC GOITRE) |
|
SECTION |
Graves’ disease, also known as Basedow’s disease, primary |
|
hyperplasia, exophthalmic goitre, and diffuse toxic goitre, is |
||
characterised by a triad of features: |
||
Hyperthyroidism (thyrotoxicosis) |
||
Diffuse thyroid enlargement |
||
III |
Ophthalmopathy. |
|
|
The disease is more frequent between the age of 30 and |
|
Systemic |
40 years and has five-fold increased prevalence among |
|
females. |
||
|
||
|
ETIOPATHOGENESIS. Graves’ disease is an autoimmune |
|
|
disease and, as already stated, there are many immunologic |
|
Pathology |
similarities between this condition and Hashimoto’s |
|
thyroiditis. These are as follows: |
||
|
||
|
1. Genetic factor association. Like in Hashimoto’s |
|
|
thyroiditis. Graves’disease too has genetic predisposition. A |
|
|
familial occurrence has been observed. Susceptibility to |
|
|
develop Graves’ disease has been found associated with |
|
|
HLA-DR3 (Hashimoto’s thyroiditis has both HLA-DR3 and |
|
|
HLA-DR5 association, page 804), CTLA-4 and PTPN22 |
|
|
(a T-cell regulatory gene). |
|
|
2. Autoimmune disease association. Graves’ disease may |
|
|
be found in association with other organ-specific auto- |
|
|
immune diseases. Hashimoto’s thyroiditis and Graves’ |
|
|
disease are frequently present in the same families and the |
|
|
two diseases may coexist in the same patient. |
|
|
3. Other factors. Besides these two factors, Graves’ disease |
|
|
has higher prevalence in women (7 to 10 times), and |
|
|
association with emotional stress and smoking. |
|
|
4. Autoantibodies. Autoantibodies against thyroid antigens |
|
|
are detectable in the serum of these patients too but their |
|
|
sites of action are different from that of Hashimoto’s |
|
|
thyroiditis. In Graves’ disease, TSH-receptor autoantigen is |
|
|
the main antigen against which autoantibodies are directed. |
|
|
These are as under: |
i)Thyroid-stimulating immunoglobulin (TSI): It binds to TSH receptor and stimulates increased release of thyroid hormone.
ii)Thyroid growth-stimulating immunoglobulins (TGI): It stimulates proliferation of follicular epithelium.
iii)TSH-binding inhibitor immunoglobulins (TBII): It is inhibitory to binding of TSH to its own receptor. Depending upon its action as inhibitory or stimulatory to follicular epithelium, it may result in alternate episodes of hypoand hyperthyroidism.
However, it is not quite clear what stimulates B cells to form these autoantibodies in Graves’ disease. Possibly, intrathyroidal CD4+ helper T cells are responsible for stimulating B cells to secrete autoantibodies.
The pathogenesis of Graves’ infiltrative ophthalmopathy is also of autoimmune origin. The evidence in support is the intense lymphocytic infiltrate around the ocular muscles and detection of circulating autoantibodies against muscle antigen that cross-react with thyroid microsomes.
MORPHOLOGIC FEATURES. Grossly, the thyroid is moderately, diffusely and symmetrically enlarged and may weigh up to 70-90 gm. On cut section, the thyroid parenchyma is typically homogeneous, red-brown and meaty and lacks the normal translucency.
Histologically, the following features are found (Fig. 27.8):
1.There is considerable epithelial hyperplasia and hypertrophy as seen by increased height of the follicular lining cells and formation of papillary infoldings of piled up epithelium into the lumina of follicles which are small.
2.The colloid is markedly diminished and is lightly staining, watery and finely vacuolated.
3.The stroma shows increased vascularity and accumulation of lymphoid cells.
However, the pathologic changes in gross specimen as well as on histologic examination are considerably altered if preoperative medication has been administered. Iodine
Figure 27.8 
 Graves’ disease. The follicles are small and are lined by tall columnar epithelium, which is piled up at places forming papillary infoldings. Colloid is nearly absent and appears lightly staining, watery and finely vacuolated.
Graves’ disease. The follicles are small and are lined by tall columnar epithelium, which is piled up at places forming papillary infoldings. Colloid is nearly absent and appears lightly staining, watery and finely vacuolated.
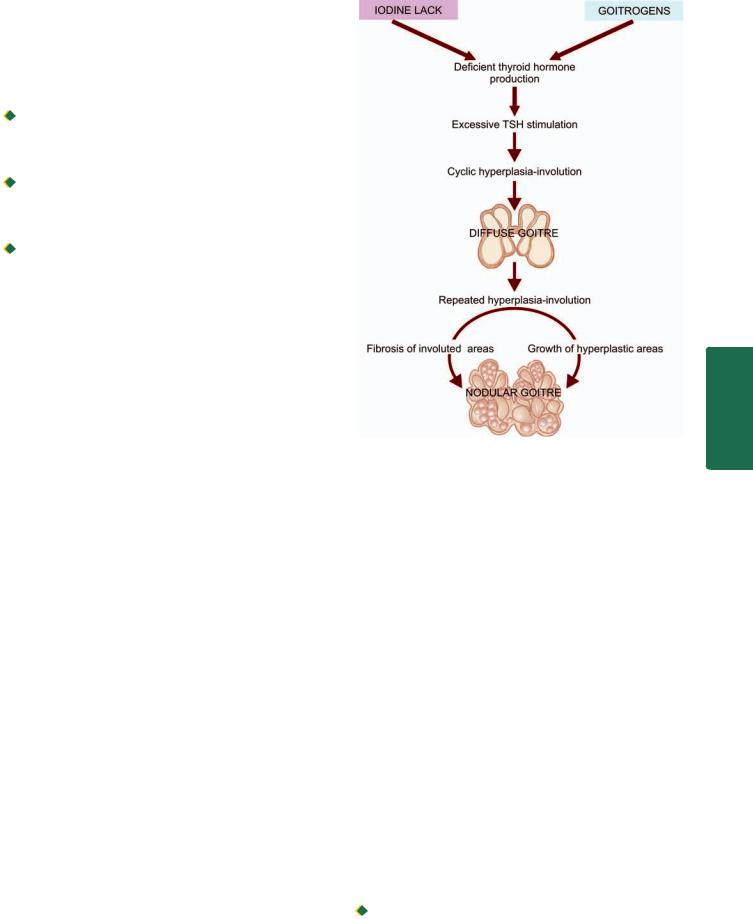
administration results in accumulation of colloid in the follicles and decrease in vascularity and height of follicular cells, while antithyroid drugs such as thiouracil cause marked hyperplasia.
CLINICAL FEATURES. Graves’ disease generally develops slowly and insidiously.
 Patients are usually young women who present with symmetric, moderate enlargement of the thyroid gland with features of thyrotoxicosis (page 802), ophthalmopathy and dermatopathy.
Patients are usually young women who present with symmetric, moderate enlargement of the thyroid gland with features of thyrotoxicosis (page 802), ophthalmopathy and dermatopathy.

 Ocular abnormalities are lid lag, upper lid retraction, stare, weakness of eye muscles and proptosis. In extreme cases, the lids can no longer close and may produce corneal injuries and ulcerations.
Ocular abnormalities are lid lag, upper lid retraction, stare, weakness of eye muscles and proptosis. In extreme cases, the lids can no longer close and may produce corneal injuries and ulcerations.
 Dermatopathy in Graves’ disease most often consists of pretibial (localised) myxoedema in the form of firm plaques.
Dermatopathy in Graves’ disease most often consists of pretibial (localised) myxoedema in the form of firm plaques.
Like in Hashimoto’s thyroiditis, there is no increased risk of development of thyroid cancer in Graves’ disease.
GOITRE
The term goitre is defined as thyroid enlargement caused by compensatory hyperplasia and hypertrophy of the follicular epithelium in response to thyroid hormone deficiency. The end-result of this hyperplasia is generally a euthyroid state (in contrast to thyrotoxicosis occurring in diffuse toxic goitre or Graves’ disease) though at some stages there may be hypoor hyperthyroidism. Two morphologic forms of goitre are distinguished:
A.Diffuse goitre (simple nontoxic goitre or colloid goitre).
B.Nodular goitre (multinodular goitre or adenomatous goitre).
Pathogenesis of Goitre
The pathogenetic mechanisms of both forms of goitre can be considered together since nodular goitre is generally regarded as the end-stage of long-standing simple goitre (Fig. 27.9). The fundamental defect is deficient production of thyroid hormones due to various etiologic factors described below, but most common is dietary lack of iodine. Deficient thyroid hormone production causes excessive TSH stimulation which leads to hyperplasia of follicular epithelium as well as formation of new thyroid follicles. Cyclical hyperplastic stage followed by involution stage completes the picture of simple goitre. Repeated and prolonged changes of hyperplasia result in continued growth of thyroid tissue while involuted areas undergo fibrosis, thus completing the picture of nodular goitre.
Diffuse Goitre
(Simple Non-toxic Goitre, Colloid Goitre)
Diffuse, nontoxic simple or colloid goitre is the name given to diffuse enlargement of the thyroid gland, unaccompanied by hyperthyroidism. Most cases are in a state of euthyroid though they may have passed through preceding stage of hypothyroidism due to inadequate supply of iodine. TSH levels are invariably elevated. In general, goitre is more
Figure 27.9 
 Pathogenesis of simple and nodular goitre.
Pathogenesis of simple and nodular goitre.
common in females. Simple goitre often appears at puberty or in adolescence, following which it may either regress or may progress to nodular goitre.
ETIOLOGY. Epidemiologically, goitre occurs in 2 forms: endemic, and non-endemic or sporadic.
Endemic goitre. Prevalence of goitre in a geographic area in more than 10% of the population is termed endemic goitre. Such endemic areas are several high mountainous regions far from the sea where iodine content of drinking water and food is low such as in the regions of the Himalayas, the Alps and the Ande. Of late, however, the prevalence in these areas has declined due to prophylactic use of iodised salt.
Though most endemic goitres are caused by dietary lack of iodine, some cases occur due to goitrogens and genetic factors. Goitrogens are substances which interfere with the synthesis of thyroid hormones. These substances are drugs used in the treatment of hyperthyroidism and certain items of food such as cabbage, cauliflower, turnips and cassava roots.
Sporadic (non-endemic) goitre. Non-endemic or sporadic simple goitre is less common than the endemic variety. In most cases, the etiology of sporadic goitre is unknown. A number of causal influences have been attributed. These include the following:

 Suboptimal iodine intake in conditions of increased demand as in puberty and pregnancy.
Suboptimal iodine intake in conditions of increased demand as in puberty and pregnancy.
807
System Endocrine The 27 CHAPTER
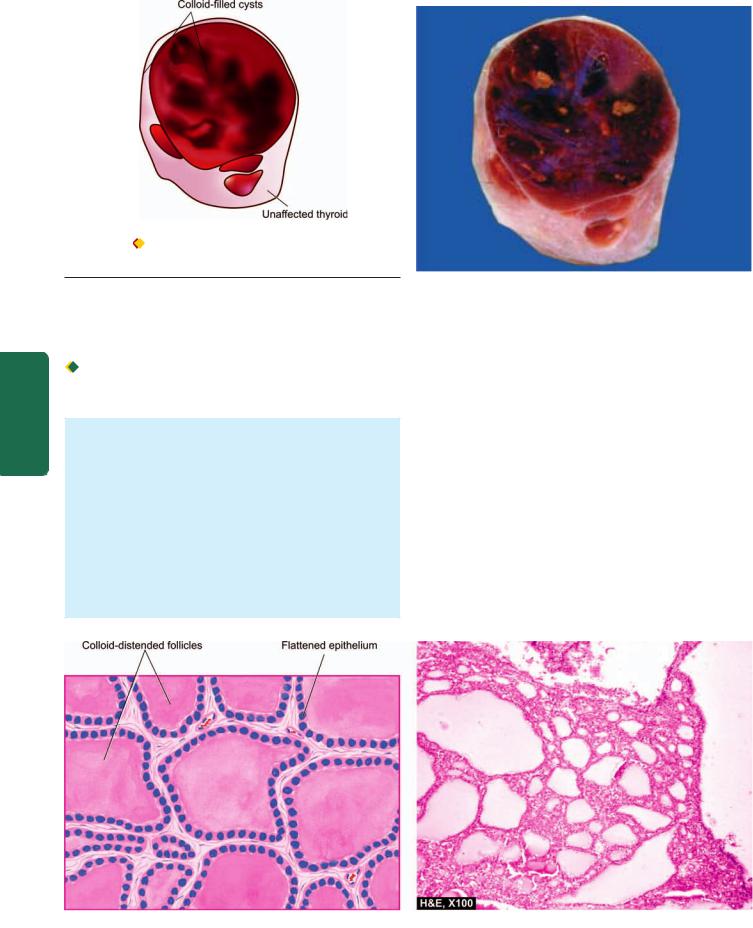
808
Figure 27.10 |
Simple (diffuse nontoxic or colloid) goitre. The thyroid |
gland is enlarged diffusely. Cut section shows lobules of translucent gelatinous light brown parenchyma and areas of haemorrhage.
Pathology Systemic III SECTION

 Genetic factors.
Genetic factors.

 Dietary goitrogenes.
Dietary goitrogenes.
 Hereditary defect in thyroid hormone synthesis and transport (dyshormonogenesis).
Hereditary defect in thyroid hormone synthesis and transport (dyshormonogenesis).

 Inborn errors of iodine metabolism.
Inborn errors of iodine metabolism.
MORPHOLOGIC FEATURES. Grossly, the enlargement of the thyroid gland in simple goitre is moderate (weighing up to 100-150 gm), symmetric and diffuse. Cut surface is gelatinous and translucent brown (Fig. 27.10).
Histologically, two stages are distinguished:
1.Hyperplastic stage is the early stage and is characterised by tall columnar follicular epithelium showing papillary infoldings and formation of small new follicles.
2.Involution stage generally follows hyperplastic stage after variable period of time. This stage is characterised by large follicles distended by colloid and lined by flattened follicular epithelium (Fig. 27.11).
Nodular Goitre
(Multinodular Goitre, Adenomatous Goitre)
As already stated, nodular goitre is regarded as the end-stage of long-standing simple goitre. It is characterised by most extreme degree of tumour-like enlargement of the thyroid gland and characteristic nodularity. The enlargement of the gland may be sufficient to not only cause cosmetic disfigurement, but in many cases may cause dsyphagia and choking due to compression of oesophagus and trachea. Most cases are in a euthyroid state but about 10% cases may develop thyrotoxicosis resulting in toxic nodular goitre or Plummer’s disease. However, thyrotoxicosis of Plummer’s disease (toxic nodular goitre) differs from that of Graves’ disease (diffuse toxic goitre) in lacking features of ophthalmopathy and dermatopathy. Such ‘hot nodules’ may be picked up by CT scan or by RAIU studies. Since nodular goitre is derived from simple goitre, it has the same female
Figure 27.11 
 Simple goitre. Microscopy shows large follicles distended by colloid and lined by flattened follicular epithelium.
Simple goitre. Microscopy shows large follicles distended by colloid and lined by flattened follicular epithelium.

809
Figure 27.12 
 Nodular goitre. The thyroid gland is enlarged and nodular. Cut surface shows multiple nodules separated from each other by incomplete fibrous septa. Areas of haemorrhage and cystic change are also seen.
Nodular goitre. The thyroid gland is enlarged and nodular. Cut surface shows multiple nodules separated from each other by incomplete fibrous septa. Areas of haemorrhage and cystic change are also seen.
preponderance but affects older individuals because it is a late complication of simple goitre.
ETIOLOGY. Etiologic factors implicated in endemic and nonendemic or sporadic variety of simple goitre are involved in the etiology of nodular goitre too. However, how nodular pattern is produced is not clearly understood. Possibly, epithelial hyperplasia, generation of new follicles, and irregular accumulation of colloid in the follicles—all contribute to produce increased tension and stress in the thyroid gland causing rupture of follicles and vessels. This is followed by haemorrhages, scarring and sometimes calcification, resulting in development of nodular pattern.
MORPHOLOGIC FEATURES. Grossly, the thyroid in nodular goitre shows asymmetric and extreme enlargement, weighing 100-500 gm or even more. The five cardinal macroscopic features are as under (Fig. 27.12):
1. Nodularity with poor encapsulation
2.Fibrous scarring
3.Haemorrhages
4.Focal calcification
5.Cystic degeneration.
Cut surface generally shows multinodularity but occasionally there may be only one or two nodules which are poorly-circumscribed (unlike complete encapsulation of thyroid adenoma, described below).
Histologically, the same heterogenicity as seen on gross appearance is seen. Corresponding microscopic features are as follows (Fig. 27.13):
1.Partial or incomplete encapsulation of nodules.
2.The follicles varying from small to large and lined by flat to high epithelium. A few may show macropapillary formation.
3.Areas of haemorrhages, haemosiderin-laden macrophages and cholesterol crystals.
4.Fibrous scarring with foci of calcification.
5.Micro-macrocystic change.
System Endocrine The 27 CHAPTER
Figure 27.13 
 Nodular goitre. The predominant histologic features are: nodularity, extensive scarring with foci of calcification, areas of haemorrhages and variable-sized follicles lined by flat to high epithelium and containing abundant colloid.
Nodular goitre. The predominant histologic features are: nodularity, extensive scarring with foci of calcification, areas of haemorrhages and variable-sized follicles lined by flat to high epithelium and containing abundant colloid.

810 TABLE 27.2: Contrasting Features of Simple and Nodular Goitre.
Feature |
Diffuse Goitre |
Nodular Goitre |
|
|
|
|
|
1. |
Nomenclature |
Simple goitre, hyperplastic goitre, nontoxic goitre |
Multinodular, adenomatous goitre |
2. |
Etiology |
Graves' disease, thyroiditis, puberty |
Endemic thyroiditis, cancer |
3. |
Pathogenesis |
Hyperplasia-involution |
Repeated cycles of hyperplasia with growth and |
|
|
|
involution with fibrosis |
4. |
Composition |
Cellular-rich |
5. |
Gross |
Moderate, symmetric, diffuse enlargement, |
|
|
colloid-filled follicles, gelatinous |
6. |
Microscopy |
Hyperplastic phase: papillary infoldings, |
|
|
Involution stage: large colloid filled |
|
|
follicles with flat epithelium |
7. |
Functional status |
Hyperthyroidism, euthyroid |
Colloid-rich
Nodular asymmetric, haemorrhages, scarring, cystic change, calcification
Incomplete encapsulation, nodularity, variable-sized follicles, fibrous scarring, haemorrhages, calcification, cyst formation
Hypothyroidism, euthyroid
Pathology Systemic III SECTION
The contrasting features of diffuse and nodular goitre are summarised in Table 27.2.
THYROID TUMOURS
Most primary tumours of the thyroid are of follicular epithelial origin; a few arise from parafollicular C-cells. The most common benign thyroid neoplasm is a follicular adenoma. Malignant tumours of the thyroid are less common but thyroid carcinoma is the most common type, though rarely lymphomas and sarcomas also occur.
FOLLICULAR ADENOMA
Follicular adenoma is the most common benign thyroid tumour occurring more frequently in adult women. Clinically, it appears as a solitary nodule which can be found in approximately 1% of the population. Besides the follicular adenoma, other conditions which may produce clinically apparent solitary nodule in the thyroid are a dominant nodule of nodular goitre and thyroid carcinoma. It is thus important to distinguish adenomas from these two conditions. Though most adenomas cause no clinical problem and behave as a ‘cold nodule’, rarely they may produce mild hyperthyroidism and appear as ‘hot nodule’ on RAIU studies. Adenoma, however, rarely ever becomes malignant.
MORPHOLOGIC FEATURES. Grossly, the follicular adenoma is characterised by four features so as to distinguish it from a nodule of nodular goitre (Fig. 27.14):
1.solitary nodule;
2.complete encapsulation;
3.clearly distinct architecture inside and outside the capsule; and
4.compression of the thyroid parenchyma outside the capsule
Usually, an adenoma is small (up to 3 cm in diameter) and spherical. On cut section, the adenoma is grey-white to red-brown, less colloidal than the surrounding thyroid parenchyma and may have degenerative changes such as fibrous scarring, focal calcification, haemorrhages and cyst formation.
Histologically, the tumour shows complete fibrous encapsulation. The tumour cells are benign follicular
epithelial cells forming follicles of various sizes or may show trabecular, solid and cord patterns with little follicle formation. Accordingly, the following 6 types of growth patterns are distinguished, though more than one pattern may be present in a single tumour:
1.Microfollicular (foetal) adenoma consists of small follicles containing little or no colloid and separated by abundant loose stroma (Fig. 27.15).
2.Normofollicular (simple) adenoma has closely packed follicles like that of normal thyroid gland.
3.Macrofollicular (colloid) adenoma contains large follicles of varying size and distended with colloid.
4.Trabecular (embryonal) adenoma resembles embryonal thyroid and consists of closely packed solid or trabecular pattern of epithelial cells with an occasional small abortive follicle.
5.Hurthle cell (oxyphilic) adenoma is an uncommon variant composed of solid trabeculae of large cells having abundant granular oxyphilic cytoplasm and vesicular
Figure 27.14 |
Follicular adenoma thyroid. Sectioned surface of the |
thyroid shows a solitary nodule having capsule. The nodule is grey-white and is distinct from the adjoining thyroid parenchyma.
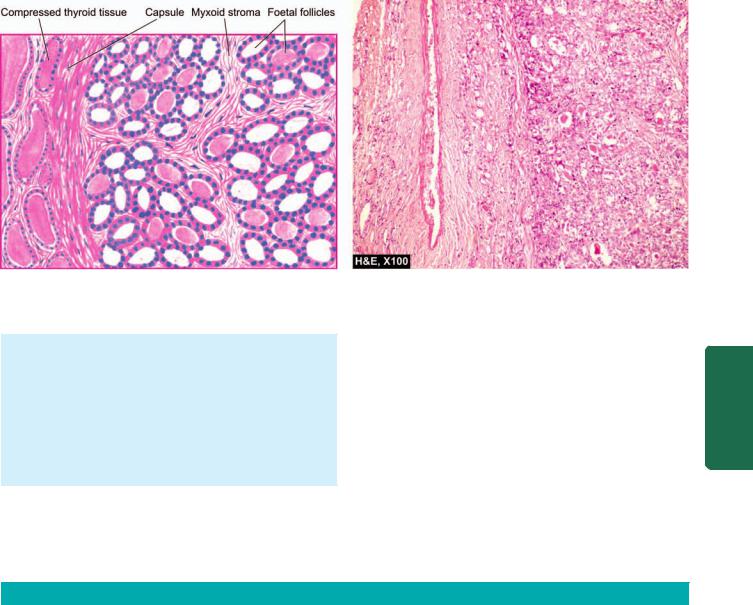
811
Figure 27.15 
 Follicular adenoma, foetal (microfollicular) type. The tumour is well-encapsulated with compression of surrounding thyroid parenchyma. The tumour consists of small follicles lined by cuboidal epithelium and contain little or no colloid and separated by abundant loose stroma.
Follicular adenoma, foetal (microfollicular) type. The tumour is well-encapsulated with compression of surrounding thyroid parenchyma. The tumour consists of small follicles lined by cuboidal epithelium and contain little or no colloid and separated by abundant loose stroma.
nuclei. The tumour cells do not form follicles and contain little stroma.
6. Atypical adenoma is the term used for a follicular adenoma which has more pronounced cellular proliferation so that features may be considered indicative of malignancy such as pleomorphism, increased mitoses and nuclear atypia. These tumours, however, do not show capsular and vascular invasion—features which distinguish it from follicular carcinoma.
THYROID CANCER
Approximately 95% of all primary thyroid cancers are carcinomas. Primary lymphomas of the thyroid comprise less than 5% of thyroid cancers and majority of them possibly
evolve from autoimmune (lymphocytic) thyroiditis (page 804). Sarcomas of the thyroid are extremely rare. About 20% of patients dying of metastasising malignancy have metastatic deposits in the thyroid gland, most commonly from malignant melanoma, renal cell carcinoma and bronchogenic carcinoma.
In line with most other thyroid lesions, most carcinomas of the thyroid too have female preponderance and are twice more common in women.
Carcinoma of the thyroid gland has 4 major morphologic types with distinctly different clinical behaviour and variable prevalence. These are: papillary, follicular, medullary and undifferentiated (anaplastic) carcinoma; their contrasting features are summed up in Table 27.3.
TABLE 27.3: Contrasting Features of Main Histologic Types of Thyroid Carcinoma.
|
Feature |
Papillary |
Follicular |
Medullary |
Anaplastic |
|
|
Carcinoma |
Carcinoma |
Carcinoma |
Carcinoma |
|
|
|
|
|
|
1. |
Frequency |
75-80% |
10-20% |
5% |
5% |
2. |
Age |
All ages |
Middle to old age |
Middle to old age; |
Old age |
|
|
|
|
familial too |
|
3. |
Female/male ratio |
3:1 |
2.5:1 |
1:1 |
1.5:1 |
4. |
Relation to radiation |
Maximum |
Present |
None |
Present |
5. |
Genetic alterations |
RET gene over- |
RAS mutation, |
RET point mutation |
p53 loss, |
|
|
expression, NTRK |
PAX-PPAR γ1 fusion |
|
β-catenin mutation |
|
|
gene rearrangement |
|
|
|
6. |
Cell of origin |
Follicular |
Follicular |
Parafollicular |
Follicular |
7. |
Gross |
Small, multifocal |
Moderate size, nodular |
Moderate size |
Invasive growth |
8. |
Pathognomonic |
Nuclear features, |
Vascular and capsular |
Solid nests, |
Undifferentiated, |
|
microscopy |
papillary pattern |
invasion |
amyloid stroma |
spindle-shaped, giant cells |
9. |
Regional metastases |
Common |
Rare |
Common |
Common |
10. |
Distant metastases |
Rare |
Common |
Rare |
Common |
11. |
10-year survival |
80-95% |
50-70% |
60-70% |
5-10% (median survival about |
|
|
|
|
|
2 months) |
|
|
|
|
|
|
System Endocrine The 27 CHAPTER
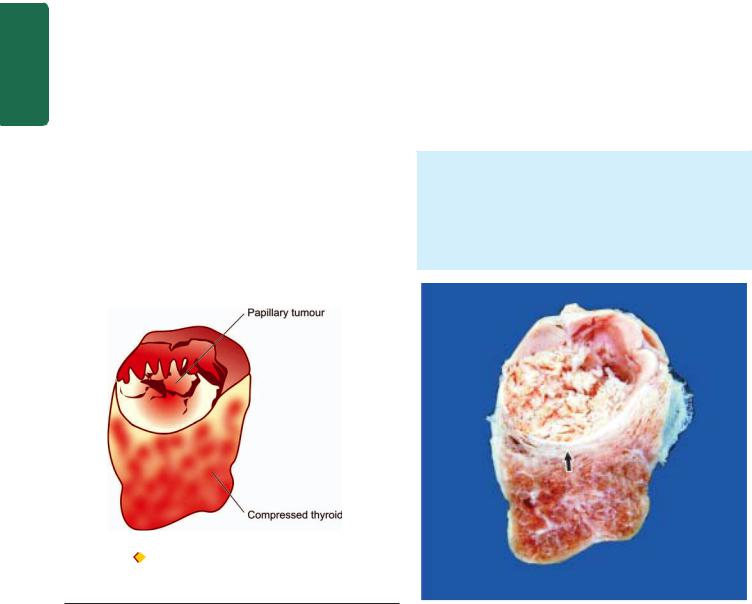
812 ETIOPATHOGENESIS. Most important risk factor implicated in the etiology of thyroid cancer is external radiation, and to a some extent there is role of TSH receptors and iodine excess, while pathogenesis of thyroid cancer is explained on genetic alterations.
1. External radiation. The single most important environmental factor associated with increased risk of developing thyroid carcinoma after many years of exposure to external radiation of high dose. Evidences in support include: high incidence of thyroid cancer in individuals irradiated in early age for enlarged thymus and for skin disorders, in Japanese atomic bomb survivors, and in individuals living in the vicinity of nuclear accident sites. In particular, exposure to radiation to children and young adults has been found to be associated with higher incidence of development of papillary carcinoma later.
|
2. Iodine excess and TSH. In regions where endemic goitre |
|
|
is widespread, addition of iodine to diet has resulted in |
|
|
increase in incidence of papillary cancer. Many well- |
|
|
differentiated thyroid cancers express TSH receptors and thus |
|
|
respond to T4 suppression of TSH. |
|
SECTION |
3. Genetic basis. Familial clustering of thyroid cancer has |
|
i) Papillary thyroid carcinoma: Mutation in RET gene (gene |
||
|
been observed, especially in medullary carcinoma. Molecular |
|
|
studies reveal that thyroid carcinoma is a multistep process |
|
|
involving genetic alterations but distinct mutations are seen |
|
|
in different histologic types: |
|
III |
overexpression) located on chromosome 10q is seen in about |
|
20% cases of papillary thyroid carcinoma. This mutation |
||
|
||
Systemic |
renders the tyrosine kinase receptor under the target of other |
|
tumour-promoting factors such as radiation exposure in |
||
|
||
|
papillary carcinoma. Another genetic abnormality seen in |
|
|
5-10% cases of papillary thyroid carcinoma is gene |
|
|
rearrangement in NTRK1 (neurotrophic tyrosine kinase |
|
Pathology |
receptor 1 located on chromosome 1q) gene. |
|
ii) Follicular thyroid carcinoma: About 50% cases of follicular |
||
|
||
|
thyroid carcinoma have mutation in RAS family of oncogenes |
|
|
that includes HRAS, NRAS and KRAS. Besides, fusion- |
translocation between 2 genes—PAX-8 (paired domain transcription factor) and PPARγ -1 (gene coding for peroxisome proliferator-activator receptor γ-1), has also been described in a proportion of cases of follicular thyroid neoplasms, both adenoma and carcinoma.
iii)Medullary thyroid carcinoma: Medullary thyroid carcinoma arises from parafollicular C-cells in the thyroid. Point mutation in RET-protooncogene is seen in both familial as a well as sporadiac cases of medullary thyroid carcinoma.
iv)Anaplastic thyroid carcinoma: This tumour either arises from further dedifferentiation of differentiated papillary or follicular thyroid carcinoma, or by inactivating point mutation in p53 tumour suppressor gene or by mutation in gene coding for β-catenin pathway.
Papillary Thyroid Carcinoma
Papillary carcinoma is the most common type of thyroid carcinoma, comprising 75-85% of cases. It can occur at all ages including children and young adults but the incidence is higher with advancing age. The tumour is found about three times more frequently in females than in males.
Papillary carcinoma is typically a slow-growing malignant tumour, most often presenting as an asymptomatic solitary nodule. Involvement of the regional lymph nodes is common but distant metastases to organs are rare. Some cases first come to attention by spread to regional lymph nodes and cause cervical lymphadenopathy. ‘Lateral aberrant thyroid’ is the term used for occurrence of thyroid tissue in the lateral cervical lymph node, which in most patients represents a well-differentiated metastasis of an occult papillary carcinoma of the thyroid.
MORPHOLOGIC FEATURES. Grossly, papillary carcinoma may range from microscopic foci to nodules upto 10 cm in diameter and is generally poorly delineated. Cut surface of the tumour is greyish-white, hard and scar-like (Fig. 27.16). Sometimes the tumour is transformed into a cyst, into which numerous papillae project and is termed
papillary cystadenocarcinoma.
Figure 27.16 |
Papillary carcinoma of the thyroid. Cut surface of the |
enlarged thyroid gland shows a single nodule separated from the rest of thyroid parenchyma by incomplete fibrous septa (arrow). The nodule is grey-white soft and shows grossly visible papillary pattern.
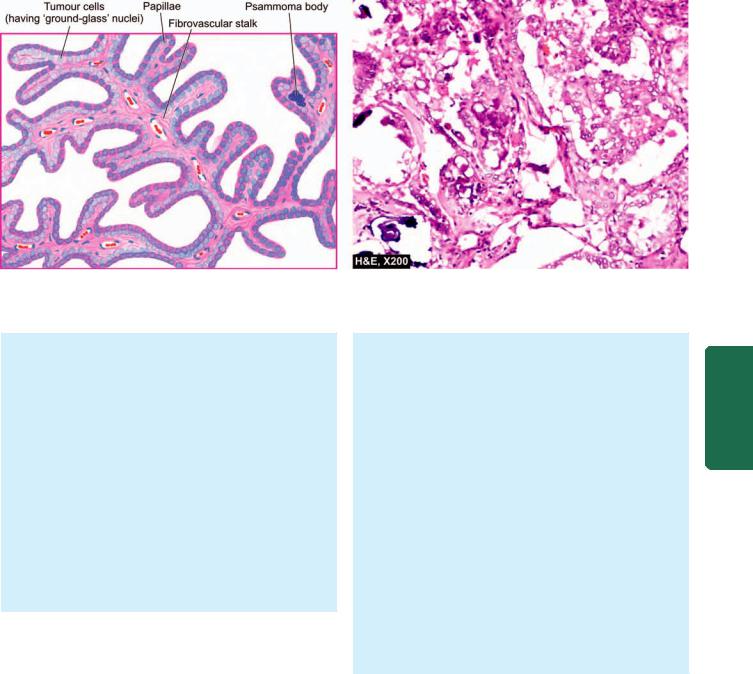
813
Figure 27.17 
 Papillary carcinoma thyroid. Microscopy shows branching papillae having flbrovascular stalk covered by a single layer of cuboidal cells having ground-glass nuclei. Colloid-filled follicles and solid sheets of tumour cells are also present.
Papillary carcinoma thyroid. Microscopy shows branching papillae having flbrovascular stalk covered by a single layer of cuboidal cells having ground-glass nuclei. Colloid-filled follicles and solid sheets of tumour cells are also present.
Histologically, the following features are present
(Fig. 27.17):
1.Papillary pattern. Papillae composed of fibrovascular stalk and covered by single layer of tumour cells is the predominant feature. Papillae are often accompanied by follicles.
2.Tumour cells. The tumour cells have characteristic nuclear features due to dispersed nuclear chromatin imparting it ground glass or optically clear appearance and clear or oxyphilic cytoplasm. These tumour cells, besides covering the papillae, may form follicles and solid sheets.
3.Invasion. The tumour cells invade the capsule and intrathyroid lymphatics but invasion of blood vessels is rare.
4.Psammoma bodies. Half of papillary carcinomas show typical small, concentric, calcified spherules called psammoma bodies in the stroma.
The prognosis of papillary carcinoma is good: 10-year survival rate is 80-95%, irrespective of whether the tumour is pure papillary or mixed papillary-follicular carcinoma.
Follicular Thyroid Carcinoma
Follicular carcinoma is the other common type of thyroid cancer, next only to papillary carcinoma and comprises about 10-20% of all thyroid carcinomas. It is more common in middle and old age and has preponderance in females (female-male ratio 2.5:1). In contrast to papillary carcinoma, follicular carcinoma has a positive correlation with endemic goitre but the role of external radiation in its etiology is unclear.
Follicular carcinoma presents clinically either as a solitary nodule or as an irregular, firm and nodular thyroid enlargement. The tumour is slow-growing but more rapid than the papillary carcinoma. In contrast to papillary carcinoma, regional lymph node metastases are rare but distant metastases by haematogenous route are common, especially to the lungs and bones.
MORPHOLOGIC FEATURES. Grossly, follicular carcinoma may be either in the form of a solitary adenomalike circumscribed nodule or as an obvious cancerous irregular thyroid enlargement. The cut surface of the tumour is grey-white with areas of haemorrhages, necrosis and cyst formation and may extend to involve adjacent structures.
Microscopically, the features are as under (Fig. 27.18):
1.Follicular pattern: Follicular carcinoma, like follicular adenoma, is composed of follicles of various sizes and may show trabecular or solid pattern. The tumour cells have hyperchromatic nuclei and the cytoplasm resembles that of normal follicular cells. However, variants like clear cell type and Hurthle cell (oxyphilic) type of follicular carcinoma may occur. The tumour differs from papillary carcinoma in lacking: papillae, ground-glass nuclei of tumour cells and psammoma bodies.
2.Vascular invasion and direct extension: Vascular invasion and direct extension to involve the adjacent structures (e.g. into the capsule) are significant features but lymphatic invasion is rare.
The prognosis of follicular carcinoma is between that of papillary and undifferentiated carcinoma: 10-year survival rate is 50-70%.
Medullary Thyroid Carcinoma
Medullary carcinoma is a less frequent type derived from parafollicular or C-cells present in the thyroid and comprises about 5% of thyroid carcinomas. It is equally common in men and women. There are 3 distinctive features which distinguish medullary carcinoma from the other thyroid carcinomas. These are: its familial occurrence, secretion of calcitonin and other peptides, and amyloid stroma.
1. Familial occurrence. Most cases of medullary carcinoma occur sporadically, but about 10% have a genetic background with point mutation in RET-protooncogene located on chromosome 10q. The familial form of medullary carcinoma
System Endocrine The 27 CHAPTER
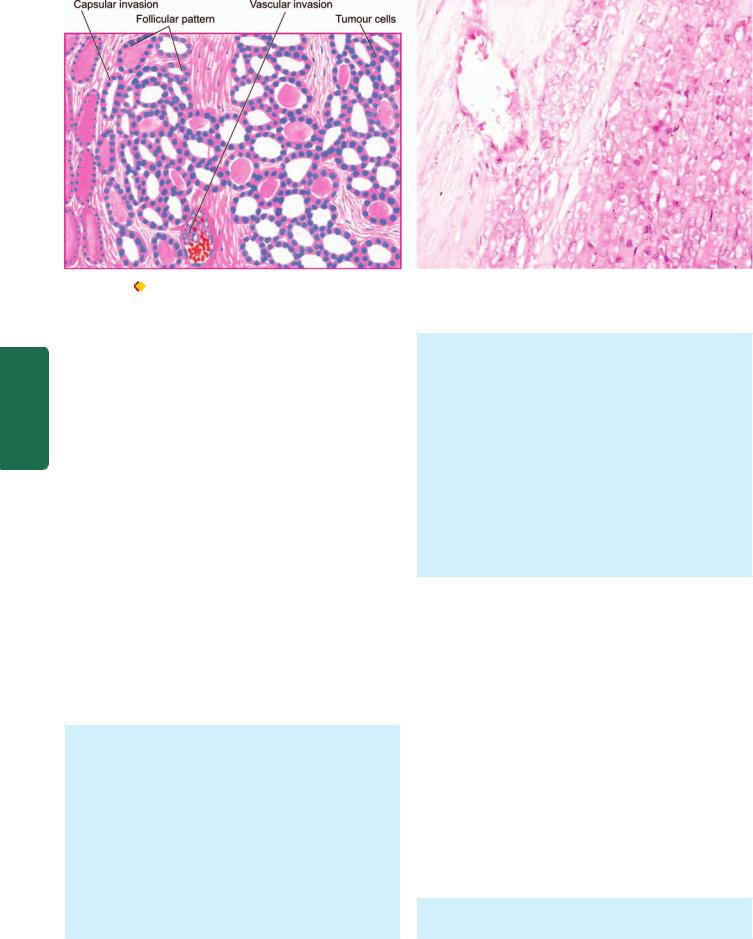
814
Pathology Systemic III SECTION
Figure 27.18 |
Follicular carcinoma, showing encapsulated tumour with invasion of a capsular vessel. The follicles lined by tumour cells are of |
various sizes and there is mild pleomorphism.
has association with pheochromocytoma and parathyroid adenoma (multiple endocrine neoplasia, MEN II A), or with pheochromocytoma and multiple mucosal neuromas (MEN II B). The sporadic cases occur in the middle and old age (5th-6th decades) and are generally unilateral, while the familial cases are found at younger age (2nd-3rd decades) and are usually bilateral and multicentric.
2.Secretion of calcitonin and other peptides. Like normal C-cells, tumour cells of medullary carcinoma secrete calcitonin, the hypocalcaemic hormone. In addition, the tumour may also elaborate prostaglandins, histaminase, somatostatin, vasoactive intestinal peptide (VIP) and ACTH. These hormone elaborations are responsible for a number of clinical syndromes such as carcinoid syndrome, Cushing’s syndrome and diarrhoea.
3.Amyloid stroma. Most medullary carcinomas have amyloid deposits in the stroma which stains positively with usual amyloid stains such as Congo red. The amyloid deposits are believed to represent stored calcitonin derived from neoplastic C-cells in the form of prohormone.
Most cases of medullary carcinoma present as solitary thyroid nodule but sometimes an enlarged cervical lymph node may be the first manifestation.
MORPHOLOGIC FEATURES. Grossly, the tumour may either appear as a unilateral solitary nodule (sporadic form), or have bilateral and multicentric involvement (familial form). However, sporadic neoplasms also eventually spread to the contralateral lobe. Cut surface of tumour in both forms shows well-defined tumour areas which are firm to hard, grey-white to yellow-brown with areas of haemorrhages and necrosis.
Histologically, the features are as under (Fig. 27.19):
1. Tumour cells: Like other neuroendocrine tumours (e.g. carcinoid, islet cell tumour, paraganglioma etc), medullary carcinoma of the thyroid too has a well-defined organoid pattern, forming nests of tumour cells separated by
fibrovascular septa. Sometimes, the tumour cells may be arranged in sheets, ribbons pseudopapillae or small follicles. The tumour cells are uniform and have the structural and functional characteristics of C-cells. Less often, the neoplastic cells are spindle-shaped.
2. Amyloid stroma: The tumour cells are separated by amyloid stroma derived from altered calcitonin which can be demonstrated by immunostain for calcitonin. The staining properties of amyloid are similar to that seen in systemic amyloidosis and may have areas of irregular calcification but without regular laminations seen in psammoma bodies.
3. C-cell hyperplasia: Familial cases generally have C-cell hyperplasia as a precursor lesion but not in sporadic cases.
Most medullary carcinomas are slow-growing. Regional lymph node metastases may occur but distant organ metastases are infrequent. The prognosis is better in familial form than in the sporadic form: overall 10-year survival rate is 60-70%.
Anaplastic Carcinoma
Undifferentiated or anaplastic carcinoma of the thyroid comprises less than 5% of all thyroid cancers and is one of the most malignant tumour in humans. The tumour is predominantly found in old age (7th-8th decades) and is slightly more common in females than in males (female-male ratio 1.5:1). The tumour is widely aggressive and rapidly growing. The features at presentation are usually those of extensive invasion of adjacent soft tissue, trachea and oesophagus. These features include: dyspnoea, dysphagia and hoarseness, in association with rapidly-growing tumour in the neck. The tumour metastasises both to regional lymph nodes and to distant organs such as the lungs.
MORPHOLOGIC FEATURES. Grossly, the tumour is generally large and irregular, often invading the adjacent
Figure 27.19  Medullary carcinoma thyroid. Microscopy shows organoid pattern of oval tumour cells and abundant amyloid stroma.
Medullary carcinoma thyroid. Microscopy shows organoid pattern of oval tumour cells and abundant amyloid stroma.
Amyloid shows congophilia which depicts apple-green birefringence under polarising microscopy.
strap muscles of the neck and other structures in the vicinity of the thyroid. Cut surface of the tumour is white and firm with areas of necrosis and haemorrhages.
Histologically, the tumour is too poorly-differentiated to be placed in any other histologic type of thyroid cancer, but usually shows a component of either papillary or follicular carcinoma in better differentiated areas. The tumour is generally composed of 3 types of cells occurring in varying proportions: small cells, spindle cells and giant cells. When one of these cell types is predominant, the histologic variant of undifferentiated carcinoma is named accordingly. Thus, there are 3 histologic variants:
1.Small cell carcinoma: This type of tumour is composed of closely packed small cells having hyperchromatic nuclei and numerous mitoses. This variant closely resembles malignant lymphoma.
2.Spindle cell carcinoma: These tumours are composed of spindle cells resembling sarcoma. Some tumours may contain obvious sarcomatous component such as areas of osteosarcoma, chondrosarcoma or rhabdomyosarcoma.
3.Giant cell carcinoma: This type is composed of highly anaplastic giant cells showing numerous atypical mitoses,
bizarre and lobed nuclei and some assuming spindle shapes.
The prognosis is poor: 5-year survival rate is less than 10% and median survival after the diagnosis is about 2 months.
PARATHYROID GLANDS
NORMAL STRUCTURE
ANATOMY. The parathyroid glands are usually 4 in number: the superior pair derived from the 3rd branchial pouch and inferior pair from the 4th branchial pouch of primitive foregut. Both pairs are usually embedded in the posterior aspect of the thyroid substance but separated from it by a connective tissue capsule. In the adults, each gland is an oval, yellowish-brown, flattened body, weighing 35-45 mg. There may, however, be variation in the number, location and size of parathyroid glands.
HISTOLOGY AND FUNCTIONS. Microscopically, parathyroid glands are composed of solid sheets and cords
815
System Endocrine The 27 CHAPTER
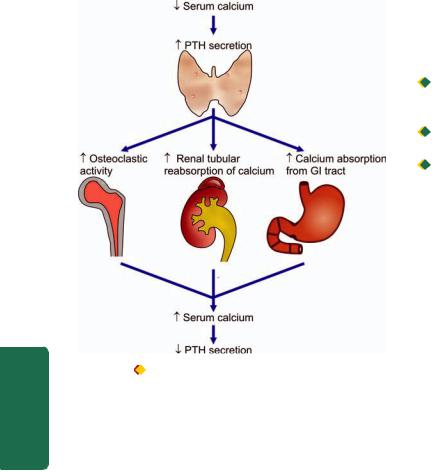
816 |
HYPERPARATHYROIDISM |
Hyperfunction of the parathyroid glands occurs due to excessive production of parathyroid hormone. It is classified into 3 types—primary, secondary and tertiary.

 Primary hyperparathyroidism occurs from oversecretion of parathyroid hormone due to disease of the parathyroid glands.
Primary hyperparathyroidism occurs from oversecretion of parathyroid hormone due to disease of the parathyroid glands.
 Secondary hyperparathyroidism is caused by diseases in other parts of the body.
Secondary hyperparathyroidism is caused by diseases in other parts of the body.

 Tertiary hyperparathyroidism develops from secondary hyperplasia after removal of the cause of secondary hyperplasia.
Tertiary hyperparathyroidism develops from secondary hyperplasia after removal of the cause of secondary hyperplasia.
Pathology Systemic III SECTION
Figure 27.20 Role of parathormone in regulating calcium metabo-
lism in the body.
of parenchymal cells and variable amount of stromal fat. The parenchymal cells are of 3 types: chief cells, oxyphil cells and water-clear cells. The chief cells are most numerous and are the major source of parathyroid hormone. The latter two types of cells appear to be derived from the chief cells and have sparse secretory granules but are potentially capable of secreting parathyroid hormone.
The major function of the parathyroid hormone, in conjunction with calcitonin and vitamin D, is to regulate serum calcium levels and metabolism of bone. Parathyroid hormone tends to elevate serum calcium level and reduce serum phosphate level. Secretion of parathyroid hormone takes place in response to serum levels of calcium by a feedback mechanism—lowered serum calcium stimulates secretion of parathyroid hormone, while elevated serum calcium causes decreased secretion of the hormone. The role of parathyroid hormone in regulating calcium metabolism in the body is at the following 3 levels (Fig. 27.20):
1.Parathyroid hormone stimulates osteoclastic activity and results in resorption of bone and release of calcium. Calcitonin released by C-cells, on the other hand, opposes parathyroid hormone by preventing resorption of bone and lowering serum calcium level.
2.Parathyroid hormone acts directly on renal tubular epithelial cells and increases renal reabsorption of calcium and inhibits reabsorption of phosphate; calcitonin enhances renal excretion of phosphate.
3.Parathyroid hormone increases renal production of the most active metabolite of vitamin D, i.e. 1, 25-dihydrocholecalci- ferol, which in turn increases calcium absorption from the small intestine.
The major parathyroid disorders are its functional disorders (hyperand hypoparathyroidism) and neoplasms.
Primary Hyperparathyroidism
Primary hyperparathyroidism is not uncommon and occurs more commonly with increasing age. It is especially likely to occur in women near the time of menopause.
ETIOLOGY. Common causes of primary hyperparathyroidism are as follows:
1.Most commonly, parathyroid adenomas in approximately 80% cases.
2.Carcinoma of the parathyroid glands in 2-3% patients.
3.Primary hyperplasia in about 15% cases (usually chief cell hyperplasia).
Also included above are the familial cases of multiple endocrine neoplasia (MEN) syndromes where parathyroid adenoma or primary hyperplasia is one of the components.
CLINICAL FEATURES. The patients with primary hyperparathyroidism have the following characteristic biochemical abnormalities:
1.Elevated levels of parathyroid hormone
2.Hypercalcaemia
3.Hypophosphataemia
4.Hypercalciuria
Clinical presentation of individuals with primary hyperparathyroidism may be in a variety of ways:
1.Most commonly, nephrolithiasis and or/nephrocalcinosis
(page 690). These dysfunctions result from excessive excretion of calcium in the urine due to hypercalcaemia induced by increased parathyroid hormone level.
2.Metastatic calcification, especially in the blood vessels, kidneys, lungs, stomach, eyes and other tissues (page 53).
3.Generalised osteitis fibrosa cystica due to osteoclastic resorption of bone and its replacement by connective tissue (page 835).
4.Neuropsychiatric disturbances such as depression, anxiety, psychosis and coma.
5.Hypertension is found in about half the cases.
6.Other changes such as pancreatitis, cholelithiasis and peptic ulcers due to hypercalcaemia and high parathyroid hormone level are less constant features.
Secondary Hyperparathyroidism
Secondary hyperparathyroidism occurs due to increased parathyroid hormone elaboration secondary to a disease
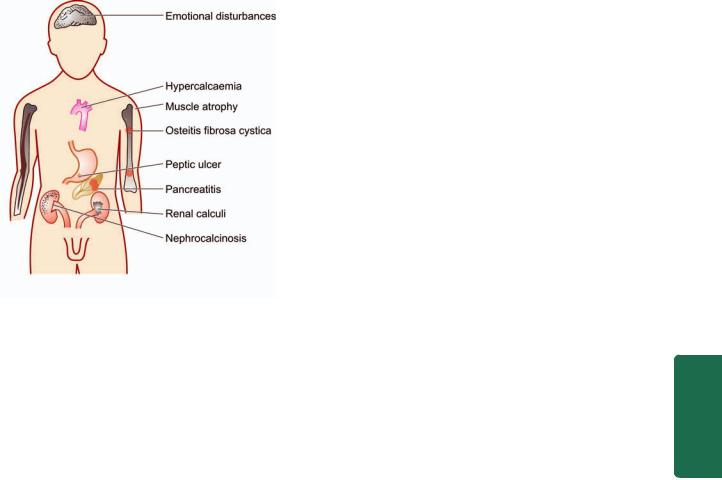
Figure 27.21 
 Major clinical manifestations of hyperparathyroidism.
Major clinical manifestations of hyperparathyroidism.
elsewhere in the body. Hypocalcaemia stimulates compensatory hyperplasia of the parathyroid glands and causes secondary hyperparathyroidism.
ETIOLOGY. Though any condition that causes hypocalcaemia stimulates excessive secretion of parathyroid hormone, the important causes of secondary hyperparathyroidism are as under:
1.Chronic renal insufficiency resulting in retention of phosphate and impaired intestinal absorption of calcium.
2.Vitamin D deficiency and consequent rickets and osteomalacia may cause parathyroid hyperfunction.
3.Intestinal malabsorption syndromes causing deficiency of calcium and vitamin D.
CLINICAL FEATURES. The main biochemical abnormality in secondary hyperparathyroidism is mild hypocalcaemia, in striking contrast to hypercalcaemia in primary hyperparathyroidism. The patients with secondary hyperparathyroidism have signs and symptoms of the disease which caused it. Usually, secondary hyperparathyroidism is a beneficial compensatory mechanism, but more severe cases may be associated with renal osteodystrophy (i.e. features of varying degree of osteitis fibrosa, osteomalacia, osteoporosis and osteosclerosis in cases of chronic renal insufficiency) and soft tissue calcification (Fig. 27.21).
Tertiary Hyperparathyroidism
Tertiary hyperparathyroidism is a complication of secondary hyperparathyroidism in which the hyperfunction persists in spite of removal of the cause of secondary hyperplasia. Possibly, a hyperplastic nodule in the parathyroid gland develops which becomes partially autonomous and continues to secrete large quantities of parathyroid hormone without regard to the needs of the body.
HYPOPARATHYROIDISM
Deficiency or absence of parathyroid hormone secretion causes hypoparathyroidism. Hypoparathyroidism is of 3
types—primary, pseudo- and pseudopseudo-hypoparathyroidism.
Primary Hypoparathyroidism |
817 |
Primary hypoparathyroidism is caused by disease of the parathyroid glands. Most common causes of primary hypoparathyroidism are: surgical procedures involving thyroid, parathyroid, or radical neck dissection for cancer. Other causes are uncommon and include idiopathic hypoparathyroidism of autoimmune origin in children and may occur as sporadic or familial cases. These cases are generally associated with other autoimmune diseases.
CLINICAL FEATURES. The main biochemical dysfunctions in primary hypoparathyroidism are hypocalcaemia, hyperphosphataemia and hypocalciuria. The clinical manifestations of these abnormalities are as under:
1.Increased neuromuscular irritability and tetany
2.Calcification of the lens and cataract formation
3.Abnormalities in cardiac conduction
4.Disorders of the CNS due to intracranial calcification
5.Abnormalities of the teeth.
Pseudo-hypoparathyroidism |
|
||
In pseudo-hypoparathyroidism, the tissues fail to respond |
|
||
to parathyroid hormone though parathyroid glands are |
CHAPTER |
||
usually normal. It is a rare inherited condition with an |
|||
|
|||
autosomal dominant character. The patients are generally |
|
||
females and are characterised by signs and symptoms of |
|
||
hypoparathyroidism and other clinical features like short |
|
||
stature, short metacarpals and metatarsals, flat nose, round |
27 |
||
face and multiple exostoses. Since renal tubules cannot |
|||
adequately respond to parathyroid hormone, there is hyper- |
|
||
calciuria, hypocalcaemia and hyperphosphataemia. |
The |
||
Pseudopseudo-hypoparathyroidism |
|||
Endocrine |
|||
Pseudopseudo-hypoparathyroidism is another rare familial |
|||
|
|||
disorder in which all the clinical features of pseudo- |
|
||
hypoparathyroidism are present except that these patients |
|
||
have no hypocalcaemia or hyperphosphataemia and the |
|
||
tissues respond normally to parathyroid hormone. |
System |
||
Pseudopseudo-hypoparathyroidism has been considered an |
|||
|
|||
incomplete form of pseudo-hypoparathyroidism. |
|
||
PARATHYROID TUMOURS |
|
||
Parathyroid adenoma and carcinoma are the neoplasms |
|
||
found in parathyroid glands, the former being much more |
|
||
common than the latter. |
|
||
Parathyroid Adenoma |
|
||
The commonest tumour of the parathyroid glands is an |
|
||
adenoma. It may occur at any age and in either sex but is |
|
||
found more frequently in adult life. Most adenomas are first |
|
||
brought to attention because of excessive secretion of |
|
||
parathyroid hormone causing features of hyperparathy- |
|
||
roidism as described above. |
|
||
|
|
|
|
MORPHOLOGIC FEATURES. Grossly, a parathyroid |
|
|
|
adenoma is small (less than 5 cm diameter) encapsulated, |
|
|
|
yellowish-brown, ovoid nodule and weighing up to 5 gm |
|
|
|
or more. |
|
|
|
|
|
|
|

818Microscopically, majority of adenomas are predominantly composed of chief cells arranged in sheets or cords. Oxyphil cells and water-clear cells may be found intermingled in varying proportions. Usually, a rim of normal parathyroid parenchyma and fat are present external to the capsule which help to distinguish an adenoma from diffuse hyperplasia.
Parathyroid Carcinoma
Carcinoma of the parathyroid is rare and produces manifestations of hyperparathyroidism which is often more pronounced. Carcinoma tends to be irregular in shape and is adherent to the adjacent tissues. Most parathyroid carcinomas are well-differentiated. It may be difficult to distinguish carcinoma of parathyroid gland from an adenoma but local invasion of adjacent tissues and distant metastases are helpful criteria of malignancy in such cases.
ENDOCRINE PANCREAS
|
The human pancreas, though anatomically a single organ, |
|
|
histologically and functionally, has 2 distinct parts—the |
|
SECTION |
exocrine and endocrine. The exocrine part of the gland and |
|
tumours. |
||
|
its disorders have already been discussed in Chapter 21. |
|
|
The discussion here is focused on the endocrine pancreas |
|
|
and its two main disorders: diabetes mellitus and islet cell |
|
III |
NORMAL STRUCTURE |
|
|
||
Systemic |
The endocrine pancreas consists of microscopic collections |
|
of cells called islets of Langerhans found scattered within |
||
the pancreatic lobules, as well as individual endocrine cells |
||
|
||
|
found in duct epithelium and among the acini. The total |
|
|
weight of endocrine pancreas in the adult, however, does |
|
Pathology |
not exceed 1-1.5 gm (total weight of pancreas 60-100 gm). |
|
The islet cell tissue is greatly concentrated in the tail than in |
||
|
||
|
the head or body of the pancreas. Islets possess no ductal |
|
|
system and they drain their secretory products directly into |
|
|
the circulation. Ultrastructurally and immunohistochemi- |
|
|
cally, 4 major and 2 minor types of islet cells are distinguished, |
|
|
each type having its distinct secretory product and function. |
|
|
These are as follows: |
A. Major cell types:
1.Beta (β) or B cells comprise about 70% of islet cells and secrete insulin, the defective response or deficient synthesis of which causes diabetes mellitus.
2.Alpha (α) or A cells comprise 20% of islet cells and secrete glucagon which induces hyperglycaemia.
3.Delta (δ) or D cells comprise 5-10% of islet cells and secrete somatostatin which suppresses both insulin and glucagon release.
4.Pancreatic polypeptide (PP) cells or F cells comprise 1-2% of islet cells and secrete pancreatic polypeptide having some gastrointestinal effects.
B. Minor cell types:
1. D1 cells elaborate vasoactive intestinal peptide (VIP) which induces glycogenolysis and hyperglycaemia and
causes secretory diarrhoea by stimulation of gastrointestinal fluid secretion.
2. Enterochromaffin cells synthesise serotonin which in pancreatic tumours may induce carcinoid syndrome.
DIABETES MELLITUS
Definition and Epidemiology
As per the WHO, diabetes mellitus (DM) is defined as a hetrogeneous metabolic disorder characterised by common feature of chronic hyperglycaemia with disturbance of carbohydrate, fat and protein metabolism.
DM is a leading cause of morbidity and mortality world over. It is estimated that approximately 1% of population suffers from DM. The incidence is rising in the developed countries of the world at the rate of about 10% per year, especially of type 2 DM, due to rising incidence of obesity and reduced activity levels. DM is expected to continue as a major health problem owing to its serious complications, especially end-stage renal disease, IHD, gangrene of the lower extremities, and blindness in the adults. It is anticipated that the number of diabetics will exceed 250 million by the year 2010.
Classification and Etiology
The older classification systems dividing DM into primary (idiopathic) and secondary types, juvenile-onset and maturity onset types, and insulin-dependent (IDDM) and non-insulin dependent (NIDDM) types, have become obsolete and undergone major revision due to extensive understanding of etiology and pathogenesis of DM in recent times.
As outlined in Table 27.4, current classification of DM based on etiology divides it into two broad categories—type
TABLE 27.4: Etiologic Classification of Diabetes Mellitus (as per American Diabetes Association, 2007).
I.TYPE 1 DIABETES MELLITUS (10%)
(earlier called Insulin-dependent, or juvenile-onset diabetes) Type IA DM: Immune-mediated
Type IB DM: Idiopathic
II.TYPE 2 DIABETES MELLITUS (80%)
(earlier called non-insulin-dependent, or maturity-onset diabetes)
III.OTHER SPECIFIC TYPES OF DIABETES (10%)
A.Genetic defect of β-cell function due to mutations in various enzymes (earlier called maturity-onset diabetes of the young or MODY) (e.g. hepatocyte nuclear transcription factor—HNF, glucokinase)
B.Genetic defect in insulin action (e.g. type A insulin resistance)
C.Diseases of exocrine pancreas (e.g. chronic pancreatitis, pancreatic tumours, post-pancreatectomy)
D.Endocrinopathies (e.g. acromegaly, Cushing's syndrome, pheochromocytoma)
E.Drugor chemical-induced (e.g. steroids, thyroid hormone, thiazides, β-blockers etc)
F.Infections (e.g. congenital rubella, cytomegalovirus)
G.Uncommon forms of immune-mediated DM (stiff man syndrome, anti-insulin receptor antibodies)
H.Other genetic syndromes (e.g. Down's syndrome, Klinefelter's syndrome, Turner's syndrome)
IV. GESTATIONAL DIABETES MELLITUS

TABLE 27.5. Major Risk Factors for Type 2 Diabetes Mellitus (ADA Recommendations, 2007).
1.Family history of type 2 DM
2.Obesity
3.Habitual physical inactivity
4.Race and ethnicity (Blacks, Asians, Pacific Islanders)
5.Previous identification of impaired fasting glucose or impaired glucose tolerance
6.History of gestational DM or delivery of baby heavier than 4 kg
7.Hypertension
8.Dyslipidaemia (HDL level < 35 mg/dl or triglycerides > 250 mg/dl)
9.Polycystic ovary disease and acanthosis nigricans
10.History of vascular disease
1 and type 2; besides there are a few uncommon specific etiologic types, and gestational DM. American Diabetes Association (2007) has identified risk factors for type 2 DM listed in Table 27.5.
Brief comments on etiologic terminologies as contrasted with former nomenclatures of DM are as under:
TYPE 1 DM. It constitutes about 10% cases of DM. It was previously termed as juvenile-onset diabetes (JOD) due to its occurrence in younger age, and was called insulindependent DM (IDDM) because it was known that these patients have absolute requirement for insulin replacement as treatment. However, in the new classification, neither age nor insulin-dependence are considered as absolute criteria. Instead, based on underlying etiology, type 1 DM is further divided into 2 subtypes:
Subtype 1A (immune-mediated) DM characterised by autoimmune destruction of β-cells which usually leads to insulin deficiency.
Subtype 1B (idiopathic) DM characterised by insulin deficiency with tendency to develop ketosis but these patients are negative for autoimmune markers.
Though type 1 DM occurs commonly in patients under 30 years of age, autoimmune destruction of β-cells can occur at any age. In fact, 5-10% patients who develop DM above 30 years of age are of type 1A DM and hence the term JOD has become obsolete.
TYPE 2 DM. This type comprises about 80% cases of DM. It was previously called maturity-onset diabetes, or non-insulin dependent diabetes mellitus (NIDDM) of obese and nonobese type.
Although type 2 DM predominantly affects older individuals, it is now known that it also occurs in obese adolescent children; hence the term MOD for it is inappropriate. Moreover, many type 2 DM patients also require insulin therapy to control hyperglycaemia or to prevent ketosis and thus are not truly non-insulin dependent contrary to its former nomenclature.
OTHER SPECIFIC ETIOLOGIC TYPES OF DM. Besides the two main types, about 10% cases of DM have a known specific etiologic defect listed in Table 27.4. One important subtype in this group is maturity-onset diabetes of the young (MODY) which has autosomal dominant inheritance, early onset of hyperglycaemia and impaired insulin secretion.
GESTATIONAL DM. About 4% pregnant women develop DM due to metabolic changes during pregnancy. Although they revert back to normal glycaemia after delivery, these women are prone to develop DM later in their life.
Pathogenesis
Depending upon etiology of DM, hyperglycaemia may result from the following:

 Reduced insulin secretion
Reduced insulin secretion

 Decreased glucose use by the body
Decreased glucose use by the body

 Increased glucose production.
Increased glucose production.
Pathogenesis of two main types of DM and its complications is distinct. In order to understand it properly, it is essential to first recall physiology of normal insulin synthesis and secretion.
NORMAL INSULIN METABOLISM. The major stimulus for both synthesis and release of insulin is glucose. The steps involved in biosynthesis, release and actions of insulin are as follows
(Fig. 27.22):
Synthesis. Insulin is synthesised in the β-cells of pancreatic islets of Langerhans:
i)It is initially formed as pre-proinsulin which is single-chain 86-amino acid precursor polypeptide.
ii)Subsequent proteolysis removes the amino terminal signal peptide, forming proinsulin.
iii)Further cleavage of proinsulin gives rise to A (21 amino acids) and B (30 amino acids) chains of insulin, linked together by connecting segment called C-peptide, all of which are stored in the secretory granules in the β-cells. As compared to A and B chains of insulin, C-peptide is less susceptible to degradation in the liver and is therefore used as a marker to distinguish endogenously synthesised and exogenously administered insulin.
For therapeutic purposes, human insulin is now produced by recombinant DNA technology.
Release. Glucose is the key regulator of insulin secretion from β-cells by a series of steps:
i)Hypoglycaemia (glucose level below 70 mg/dl or below 3.9 mmol/L) stimulates transport into β-cells of a glucose transporter, GLUT2. Other stimuli influencing insulin release include nutrients in the meal, ketones, amino acids etc.
ii)An islet transcription factor, glucokinase, causes glucose phosphorylation, and thus acts as a step for controlled release of glucose-regulated insulin secretion.
iii)Metabolism of glucose to glucose-6-phosphate by glycolysis generates ATP.
iv)Generation of ATP alters the ion channel activity on the membrane. It causes inhibition of ATP-sensitive K+ channel on the cell membrane and opening up of calcium channel with resultant influx of calcium, which stimulates insulin release.
Action. Half of insulin secreted from β-cells into portal vein is degraded in the liver while the remaining half enters the systemic circulation for action on the target cells:
i)Insulin from circulation binds to its receptor on the target cells. Insulin receptor has intrinsic tyrosine kinase activity.
ii)This, in turn, activates post-receptor intracellular signalling pathway molecules, insulin receptor substrates (IRS) 1 and 2
819
System Endocrine The 27 CHAPTER
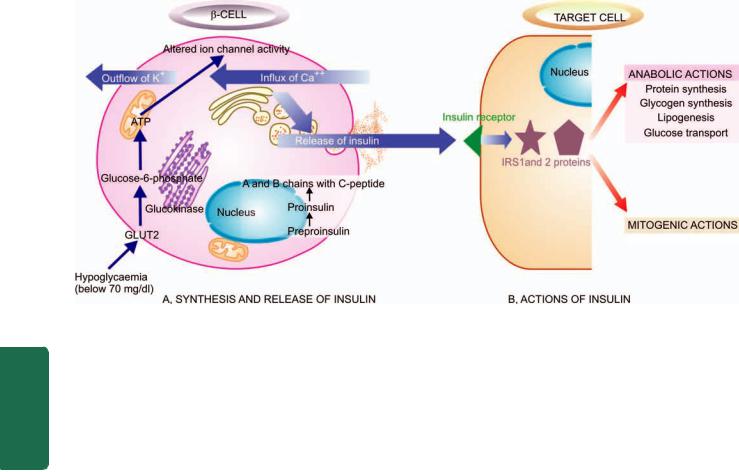
820
Pathology Systemic III SECTION
Figure 27.22 
 A, Pathway of normal insulin synthesis and release in β-cells of pancreatic islets. B, Chain of events in action of insulin on target cell.
A, Pathway of normal insulin synthesis and release in β-cells of pancreatic islets. B, Chain of events in action of insulin on target cell.
proteins, which initiate sequence of phosphorylation and dephosphorylation reactions.
iii)These reactions on the target cells are responsible for the main mitogenic and anabolic actions of insulin—glycogen synthesis, glucose transport, protein synthesis, lipogenesis.
iv)Besides the role of glucose in maintaining equilibrium of insulin release, low insulin level in the fasting state promotes hepatic gluconeogenesis and glycogenolysis, reduced glucose uptake by insulin-sensitive tissues and promotes mobilisation of stored precursors, so as to prevent hypoglycaemia.
PATHOGENESIS OF TYPE 1 DM. The basic phenomenon in
type 1 DM is destruction of β-cell mass, usually leading to absolute insulin deficiency. While type 1B DM remains idiopathic, pathogenesis of type 1A DM is immune-mediated and has been extensively studied. Currently, pathogenesis of type 1A DM is explained on the basis of 3 mutually-interlinked mechanisms: genetic susceptibility, autoimmune factors, and certain environmental factors (Fig. 27.23,A).
1. Genetic susceptibility. Type 1A DM involves inheritance of multiple genes to confer susceptibility to the disorder:
i)It has been observed in identical twins that if one twin has type 1A DM, there is about 50% chance of the second twin developing it, but not all. This means that some additional modifying factors are involved in development of DM in these cases.
ii)About half the cases with genetic predisposition to type 1A DM have the susceptibility gene located in the HLA region of chromosome 6 (MHC class II region), particularly HLA DR3, HLA DR4 and HLA DQ locus.
2. Autoimmune factors. Studies on humans and animal models on type 1A DM have shown several immunologic abnormalities:
i) Presence of islet cell antibodies against GAD (glutamic acid decarboxylase), insulin etc, though their assay largely remains a research tool due to tedious method.
ii)Occurrence of lymphocytic infiltrate in and around the pancreatic islets termed insulitis. It chiefly consists of CD8+ T lymphocytes with variable number of CD4+ T lymphocytes and macrophages.
iii)Selective destruction of β-cells while other islet cell types (glucagon-producing alpha cells, somatostatin-producing delta cells, or polypeptide-forming PP cells) remain unaffected. This is mediated by T-cell mediated cytotoxicity or by apoptosis.
iv)Role of T cell-mediated autoimmunity is further supported by transfer of type 1A DM from diseased animal by infusing T lymphocytes to a healthy animal.
v)Association of type 1A DM with other autoimmune diseases in about 10-20% cases such as Graves’ disease, Addison’s disease, Hashimoto’s thyroiditis, pernicious anaemia.
vi)Remission of type 1A DM in response to immunosuppressive therapy such as administration of cyclosporin A.
3. Environmental factors. Epidemiologic studies in type 1A DM suggest the involvement of certain environmental factors in its pathogenesis, though role of none of them has been conclusively proved. In fact, the trigger may precede the occurrence of the disease by several years. It appears that certain viral and dietary proteins share antigenic properties with human cell surface proteins and trigger the immune attack on β-cells by a process of molecular mimicry. These factors include the following:
i)Certain viral infections preceding the onset of disease e.g. mumps, measles, coxsackie B virus, cytomegalovirus and infectious mononucleosis.
ii)Experimental induction of type 1A DM with certain chemicals has been possible e.g. alloxan, streptozotocin and pentamidine.
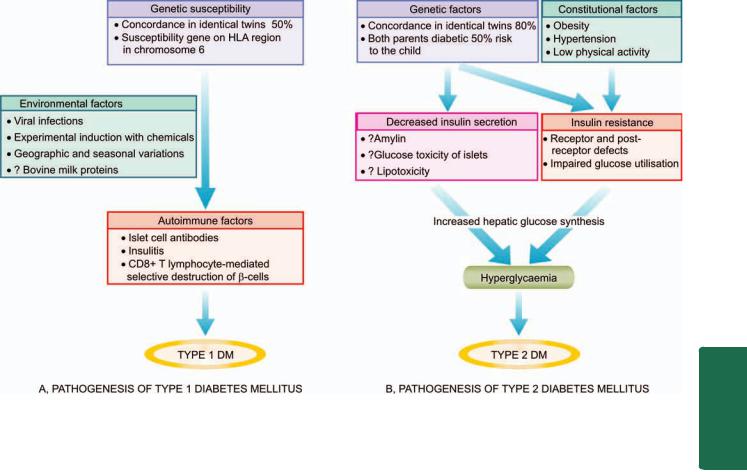
Figure 27.23 
 Schematic mechanisms involved in pathogenesis of two main types of diabetes mellitus.
Schematic mechanisms involved in pathogenesis of two main types of diabetes mellitus.
iii)Geographic and seasonal variations in its incidence suggest some common environmental factors.
iv)Possible relationship of early exposure to bovine milk proteins and occurrence of autoimmune process in type 1A DM is being studied.
KEY POINTS: Pathogenesis of type 1A DM can be summed up by interlinking the above three factors as under:
1.At birth, individuals with genetic susceptibility to this disorder have normal β-cell mass.
2.β-cells act as autoantigens and activate CD4+ T lymphocytes, bringing about immune destruction of pancreatic β-cells by autoimmune phenomena and takes months to years. Clinical features of diabetes manifest after more than 80% of β-cell mass has been destroyed.
3.The trigger for autoimmune process appears to be some infectious or environmental factor which specifically targets
β-cells.
PATHOGENESIS OF TYPE 2 DM. The basic metabolic defect in type 2 DM is either a delayed insulin secretion relative to glucose load (impaired insulin secretion), or the peripheral tissues are unable to respond to insulin (insulin
resistance).
Type 2 DM is a heterogeneous disorder with a more complex etiology and is far more common than type 1, but much less is known about its pathogenesis. A number of factors have been implicated though, but HLA association and autoimmune phenomena are not implicated. These factors are as under (Fig. 27.23,B):
1.Genetic factors. Genetic component has a stronger basis for type 2 DM than type 1A DM. Although no definite and consistent genes have been identified, multifactorial inheritance is the most important factor in development of type 2 DM:
i)There is approximately 80% chance of developing diabetes in the other identical twin if one twin has the disease.
ii)A person with one parent having type 2 DM is at an increased risk of getting diabetes, but if both parents have type
2DM the risk in the offspring rises to 40%.
2.Constitutional factors. Certain environmental factors such as obesity, hypertension, and level of physical activity play contributory role and modulate the phenotyping of the disease.
3.Insulin resistance. One of the most prominent metabolic features of type 2 DM is the lack of responsiveness of peripheral tissues to insulin, especially of the skeletal muscle and liver. Obesity, in particular, is strongly associated with insulin resistance and hence type 2 DM. Mechanism of hyperglycaemia in these cases is explained as under:
i)Resistance to action of insulin impairs glucose utilisation and hence hyperglycaemia.
ii)There is increased hepatic synthesis of glucose.
iii)Hyperglycaemia in obesity is related to high levels of free fatty acids and cytokines (e.g. TNF-α and adiponectin) affect peripheral tissue sensitivity to respond to insulin.
The precise underlying molecular defect responsible for insulin resistance in type 2 DM has yet not been fully
821
System Endocrine The 27 CHAPTER
822identified. Currently, it is proposed that insulin resistance may be possibly due to one of the following defects:

 Polymorphism in various post-receptor intracellular signal
Polymorphism in various post-receptor intracellular signal
|
pathway molecules. |
|
|
Elevated free fatty acids seen in obesity may contribute e.g. |
|
|
by impaired glucose utilisation in the skeletal muscle, by |
|
|
increased hepatic synthesis of glucose, and by impaired |
|
|
β-cell function. |
|
|
Insulin resistance syndrome is a complex of clinical features |
|
|
occurring from insulin resistance and its resultant metabolic |
|
|
derangements that includes hyperglycaemia and |
|
|
compensatory hyperinsulinaemia. The clinical features are |
|
|
in the form of accelerated cardiovascular disease and may |
|
|
occur in both obese as well as non-obese type 2 DM patients. |
|
|
The features include: mild hypertension (related to |
|
|
endothelial dysfunction) and dyslipidaemia (characterised |
|
|
by reduced HDL level, increased triglycerides and LDL level). |
|
|
4. Impaired insulin secretion. In type 2 DM, insulin |
|
|
resistance and insulin secretion are interlinked: |
|
|
i) Early in the course of disease, in response to insulin |
|
|
resistance there is compensatory increased secretion of |
|
SECTION |
insulin (hyperinsulinaemia) in an attempt to maintain normal |
|
blood glucose level. |
||
|
||
|
ii)Eventually, however, there is failure of β-cell function to |
|
|
secrete adequate insulin, although there is some secretion of |
|
|
insulin i.e. cases of type 2 DM have mild to moderate |
|
III |
deficiency of insulin (which is much less severe than that in |
|
type 1 DM) but not its total absence. |
||
|
||
|
The exact genetic mechanism why there is a fall in insulin |
|
Systemic |
secretion in these cases is unclear. However, following |
|
possibilities are proposed: |
||
|
||
|
Islet amyloid polypeptide (amylin) which forms fibrillar |
|
|
protein deposits in pancreatic islets in longstanding cases of |
|
Pathology |
type 2 DM may be responsible for impaired function of |
|
β-cells of islet cells. |
||
|
||
|
Metabolic environment of chronic hyperglycaemia |
|
|
surrounding the islets (glucose toxicity) may paradoxically |
|
|
impair islet cell function. |
|
|
Elevated free fatty acid levels (lipotoxicity) in these cases |
|
|
may worsen islet cell function. |
|
|
5. Increased hepatic glucose synthesis. One of the normal |
|
|
roles played by insulin is to promote hepatic storage of |
|
|
glucose as glycogen and suppress gluconeogenesis. In type |
|
|
2 DM, as a part of insulin resistance by peripheral tissues, |
|
|
the liver also shows insulin resistance i.e. in spite of hyper- |
|
|
insulinaemia in the early stage of disease, gluconeogenesis |
|
|
in the liver is not suppressed. This results in increased hepatic |
|
|
synthesis of glucose which contributes to hyperglycaemia |
|
|
in these cases. |
KEY POINTS: In essence, hyperglycaemia in type 2 DM is not due to destruction of β-cells but is instead a failure of β- cells to meet the requirement of insulin in the body. Its pathogenesis can be summed up by interlinking the above factors as under:
1.Type 2 DM is a more complex multifactorial disease.
2.There is greater role of genetic defect and heredity.
3.Two main mechanisms for hyperglycaemia in type 2 DM— insulin resistance and impaired insulin secretion, are interlinked.
4.While obesity plays a role in pathogenesis of insulin resistance, impaired insulin secretion may be from many constitutional factors.
5.Increased hepatic synthesis of glucose in initial period of disease contributes to hyperglycaemia.
Morphologic Features in Pancreatic Islets
Morphologic changes in islets have been demonstrated in both types of diabetes, though the changes are more distinctive in type 1 DM:
1. Insulitis:
In type 1 DM, characteristically, in early stage there is lymphocytic infiltrate, mainly by T cells, in the islets which may be accompanied by a few macrophages and polymorphs. Diabetic infants born to diabetic mothers, however, have eosinophilic infiltrate in the islets.
In type 2 DM, there is no significant leucocytic infiltrate in the islets but there is variable degree of fibrous tissue in the islets.
2. Islet cell mass:
In type 1 DM, as the disease becomes chronic there is progressive depletion of β−cell mass, eventually resulting in total loss of pancreatic β−cells and its hyalinisation.
In type 2 DM, β-cell mass is either normal or mildly reduced. Infants of diabetic mothers, however, have hyperplasia and hypertrophy of islets as a compensatory response to maternal hyperglycaemia.
3. Amyloidosis:
In type 1 DM, deposits of amyloid around islets are absent.
In type 2 DM, characteristically chronic long-standing cases show deposition of amyloid material, amylin, around the capillaries of the islets causing compression and atrophy of islet tissue (Fig. 27.24).
Figure 27.24 
 Amyloidosis of the pancreatic islet tissue. The islets are mostly replaced by structureless eosinophilic material which stains positively with Congo red.
Amyloidosis of the pancreatic islet tissue. The islets are mostly replaced by structureless eosinophilic material which stains positively with Congo red.
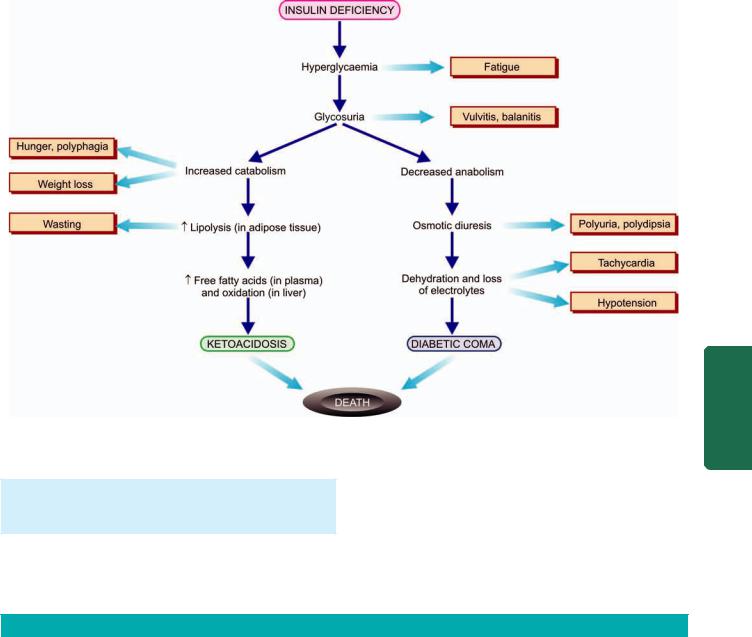
Figure 27.25 
 Pathophysiological basis of common signs and symptoms due to uncontrolled hyperglycaemia in diabetes mellitus.
Pathophysiological basis of common signs and symptoms due to uncontrolled hyperglycaemia in diabetes mellitus.
4. β-cell degranulation: In type 1 DM, EM shows degranulation of remaining β-cells of islets.
In type 2 DM, no such change is observed.
Clinical Features
It can be appreciated that hyperglycaemia in DM does not cause a single disease but is associated with numerous
diseases and symptoms, especially due to complications. Two main types of DM can be distinguished clinically to the extent shown in Table 27.6. However, overlapping of clinical features occurs as regards the age of onset, duration of symptoms and family history. Pathophysiology in evolution of clinical features is schematically shown in
Fig. 27.25.
TABLE 27.6: Contrasting Features of Type 1 and Type 2 Diabetes Mellitus.
|
Feature |
Type 1 DM |
Type 2 DM |
|
|
|
|
1. |
Frequency |
10-20% |
80-90% |
2. |
Age at onset |
Early (below 35 years) |
Late (after 40 years) |
3. |
Type of onset |
Abrupt and severe |
Gradual and insidious |
4. |
Weight |
Normal |
Obese/non-obese |
5. |
HLA |
Linked to HLA DR3, HLA DR4, HLA DQ |
No HLA association |
6. |
Family history |
< 20% |
About 60% |
7. |
Genetic locus |
Unknown |
Chromosome 6 |
8. |
Diabetes in identical twins |
50% concordance |
80% concordance |
9. |
Pathogenesis |
Autoimmune destruction of β-cells |
Insulin resistance, impaired insulin |
|
|
|
secretion |
10. |
Islet cell antibodies |
Yes |
No |
11. |
Blood insulin level |
Decreased insulin |
Normal or increased insulin |
12. |
Islet cell changes |
Insulitis, β-cell depletion |
No insulitis, later fibrosis of islets |
13. |
Amyloidosis |
Infrequent |
Common in chronic cases |
14. |
Clinical management |
Insulin and diet |
Diet, exercise, oral drugs, insulin |
15. |
Acute complications |
Ketoacidosis |
Hyperosmolar coma |
|
|
|
|
823
System Endocrine The 27 CHAPTER
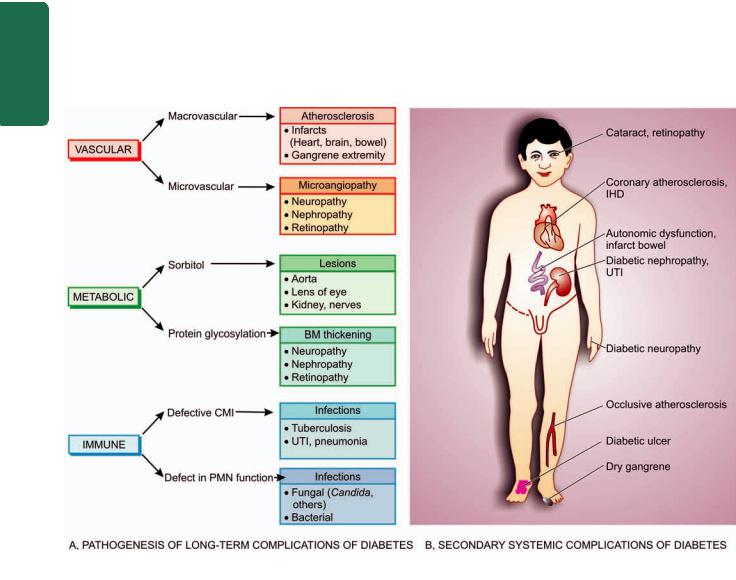
824Type 1 DM:
i)Patients of type 1 DM usually manifest at early age, generally below the age of 35.
ii)The onset of symptoms is often abrupt.
iii)At presentation, these patients have polyuria, polydipsia and polyphagia.
iv)The patients are not obese but have generally progressive loss of weight.
v)These patients are prone to develop metabolic complications such as ketoacidosis and hypoglycaemic episodes.
|
Type 2 DM: |
|
|
i) This form of diabetes generally manifests in middle life |
|
|
or beyond, usually above the age of 40. |
|
|
ii) The onset of symptoms in type 2 DM is slow and |
|
|
insidious. |
|
|
iii) Generally, the patient is asymptomatic when the |
|
|
diagnosis is made on the basis of glucosuria or hyper- |
|
|
glycaemia during physical examination, or may present with |
|
|
polyuria and polydipsia. |
|
IIISECTION |
iv) The patients are frequently obese and have unexplained |
|
weakness and loss of weight. |
||
|
||
|
v) Metabolic complications such as ketoacidosis are |
|
|
infrequent. |
|
Pathology Systemic |
|
Pathogenesis of Complications
It is now known that in both type 1 and 2 DM, severity and chronicity of hyperglycaemia forms the main pathogenetic mechanism for ‘microvascular complications’ (e.g. retinopathy, nephropathy, neuropathy); therefore control of blood glucose level constitutes the mainstay of treatment for minimising development of these complications. Longstanding cases of type 2 DM, however, in addition, frequently develop ‘macrovascular complications’ (e.g. atherosclerosis, coronary artery disease, peripheral vascular disease, cerebrovascular disease) which are more difficult to explain on the basis of hyperglycaemia alone.
The following biochemical mechanisms have been proposed to explain the development of complications of diabetes mellitus (Fig. 27.26, A):
1. Non-enzymatic protein glycosylation: The free amino group of various body proteins binds by non-enzymatic mechanism to glucose; this process is called glycosylation and is directly proportionate to the severity of hyperglycaemia. Various body proteins undergoing chemical alterations in this way include haemoglobin, lens crystalline protein, and basement membrane of body cells. An example is the measurement of a fraction of haemoglobin called glycosylated haemoglobin (HbA1C) as a test for monitoring glycaemic control in a diabetic patient during the preceding
Figure 27.26 
 Long-term complications of diabetes mellitus. A, Pathogenesis. B, Secondary systemic complications.
Long-term complications of diabetes mellitus. A, Pathogenesis. B, Secondary systemic complications.
90 to 120 days which is lifespan of red cells (page 288) . Similarly, there is accumulation of labile and reversible glycosylation products on collagen and other tissues of the blood vessel wall which subsequently become stable and irreversible by chemical changes and form advanced glycosylation end-products (AGE). The AGEs bind to receptors on different cells and produce a variety of biologic and chemical changes e.g. thickening of vascular basement membrane in diabetes.
2. Polyol pathway mechanism. This mechanism is responsible for producing lesions in the aorta, lens of the eye, kidney and peripheral nerves. These tissues have an enzyme, aldose reductase, that reacts with glucose to form sorbitol and fructose in the cells of the hyperglycaemic patient as under:
|
aldose reductase |
||
Glucose + NADH + H+ |
> |
Sorbitol + NAD+ |
|
|
sorbitol |
|
|
Sorbitol + NAD |
dehydrogenase |
> Fructose + NADH + H+ |
|
|
|||
Intracellular accumulation of sorbitol and fructose so produced results in entry of water inside the cell and consequent cellular swelling and cell damage. Also, intracellular accumulation of sorbitol causes intracellular deficiency of myoinositol which promotes injury to Schwann cells and retinal pericytes. These polyols result in disturbed processing of normal intermediary metabolites leading to complications of diabetes.
3. Excessive oxygen free radicals. In hyperglycaemia, there is increased production of reactive oxygen free radicals from mitochondrial oxidative phosphorylation which may damage various target cells in diabetes.
Complications of Diabetes
As a consequence of hyperglycaemia of diabetes, every tissue and organ of the body undergoes biochemical and structural alterations which account for the major complications in diabetics which may be acute metabolic or chronic systemic.
Both types of diabetes mellitus may develop complications which are broadly divided into 2 major groups:
I. Acute metabolic complications: These include diabetic ketoacidosis, hyperosmolar nonketotic coma, and hypoglycaemia.
II. Late systemic complications: These are atherosclerosis, diabetic microangiopathy, diabetic nephropathy, diabetic neuropathy, diabetic retinopathy and infections.
I. ACUTE METABOLIC COMPLICATIONS. Metabolic complications develop acutely. While ketoacidosis and hypoglycaemic episodes are primarily complications of type 1 DM, hyperosmolar nonketotic coma is chiefly a complication of type 2 DM (also see Fig. 27.25).
1. Diabetic ketoacidosis (DKA). Ketoacidosis is almost exclusively a complication of type 1 DM. It can develop in patients with severe insulin deficiency combined with glucagon excess. Failure to take insulin and exposure to stress
are the usual precipitating causes. Severe lack of insulin causes lipolysis in the adipose tissues, resulting in release of free fatty acids into the plasma. These free fatty acids are taken up by the liver where they are oxidised through acetyl coenzyme-A to ketone bodies, principally acetoacetic acid and β-hydroxybutyric acid. Such free fatty acid oxidation to ketone bodies is accelerated in the presence of elevated level of glucagon. Once the rate of ketogenesis exceeds the rate at which the ketone bodies can be utilised by the muscles and other tissues, ketonaemia and ketonuria occur. If urinary excretion of ketone bodies is prevented due to dehydration, systemic metabolic ketoacidosis occurs. Clinically, the condition is characterised by anorexia, nausea, vomitings, deep and fast breathing, mental confusion and coma. Most patients of ketoacidosis recover.
2.Hyperosmolar hyperglycaemic nonketotic coma (HHS).
Hyperosmolar hyperglycaemic nonketotic coma is usually a complication of type 2 DM. It is caused by severe dehydration resulting from sustained hyperglycaemic diuresis. The loss of glucose in urine is so intense that the patient is unable to drink sufficient water to maintain urinary fluid loss. The usual clinical features of ketoacidosis are absent but prominent central nervous signs are present. Blood sugar is extremely high and plasma osmolality is high. Thrombotic and bleeding complications are frequent due to high viscosity of blood. The mortality rate in hyperosmolar nonketotic coma is high.
The contrasting features of diabetic ketoacidosis and hyperosmolar non-ketotic coma are summarised in
Table 27.7.
3.Hypoglycaemia. Hypoglycaemic episode may develop in patients of type 1 DM. It may result from excessive administration of insulin, missing a meal, or due to stress. Hypoglycaemic episodes are harmful as they produce permanent brain damage, or may result in worsening of diabetic control and rebound hyperglycaemia, so called
Somogyi’s effect.
II. LATE SYSTEMIC COMPLICATIONS. A number of systemic complications may develop after a period of
TABLE 27.7: Contrasting Features of Diabetic Ketoacidosis (DKA) and Hyperosmolar Hyperglycaemic Non-ketotic Coma (HHS).
|
Lab Findings |
DKA |
HHS |
i. |
Plasma glucose (mg/dL) |
250-600 |
> 600 |
ii. |
Plasma acetone |
+ |
Less + |
iii. |
S. Na+ (mEq/L) |
Usually low |
N, ↑ or low |
iv. |
S. K+ (mEq/L) |
N, ↑ or low |
N or ↑ |
v. |
S. phosphorus (mEq/L) |
N or ↑ |
N or ↑ |
vi. |
S. Mg++ |
N or ↑ |
N or ↑ |
vii. |
S. bicarbonate (mEq/L) |
Usually <15 |
Usually >20 |
viii. |
Blood pH |
<7.30 |
> 7.30 |
ix. |
S. osmolarity (mOsm/L) |
<320 |
> 330 |
x. |
S. lactate (mmol/L) |
2-3 |
1-2 |
xi. |
S. BUN (mg/dL) |
Less ↑ |
Greater ↑ |
xii. |
Plasma insulin |
Low to 0 |
Some |
|
|
|
|
825
System Endocrine The 27 CHAPTER

826 15-20 years in either type of diabetes. Late complications are largely responsible for morbidity and premature mortality in diabetes mellitus. These complications are briefly outlined below as they are discussed in detail in relevant chapters
(Fig. 27.26,B).
1. Atherosclerosis. Diabetes mellitus of both type 1 and type 2 accelerates the development of atherosclerosis. Consequently, atherosclerotic lesions appear earlier than in the general population, are more extensive, and are more often associated with complicated plaques such as ulceration, calcification and thrombosis (page 398). The cause for this accelerated atherosclerotic process is not known but possible contributory factors are hyperlipidaemia, reduced HDL levels, nonenzymatic glycosylation, increased platelet adhesiveness, obesity and associated hypertension in diabetes.
The possible ill-effects of accelerated atherosclerosis in diabetes are early onset of coronary artery disease, silent myocardial infarction, cerebral stroke and gangrene of the toes and feet. Gangrene of the lower extremities is 100 times
|
more common in diabetics than in non-diabetics. |
|
SECTION |
2. Diabetic microangiopathy. Microangiopathy of diabetes |
|
is characterised by basement membrane thickening of small |
||
blood vessels and capillaries of different organs and tissues |
||
such as the skin, skeletal muscle, eye and kidney. Similar |
||
type of basement membrane-like material is also deposited |
||
in nonvascular tissues such as peripheral nerves, renal |
||
III |
tubules and Bowman’s capsule. The pathogenesis of diabetic |
|
microangiopathy as well as of peripheral neuropathy in dia- |
||
|
||
Systemic |
betics is believed to be due to recurrent hyperglycaemia that |
|
3. Diabetic nephropathy. Renal involvement is a common |
||
|
causes increased glycosylation of haemoglobin and other |
|
|
proteins (e.g. collagen and basement membrane material) |
|
|
resulting in thickening of basement membrane. |
|
Pathology |
complication and a leading cause of death in diabetes. Four |
|
nodular lesions of glomerulosclerosis. |
types of lesions are described in diabetic nephropathy (page 677):
i) Diabetic glomerulosclerosis which includes diffuse and
ii) Vascular lesions that include hyaline arteriolosclerosis of afferent and efferent arterioles and atheromas of renal arteries.
iii) Diabetic pyelonephritis and necrotising renal papillitis. iv) Tubular lesions or Armanni-Ebstein lesion.
4. Diabetic neuropathy. Diabetic neuropathy may affect all parts of the nervous system but symmetric peripheral neuropathy is most characteristic. The basic pathologic changes are segmental demyelination, Schwann cell injury and axonal damage (page 892). The pathogenesis of neuropathy is not clear but it may be related to diffuse microangiopathy as already explained, or may be due to accumulation of sorbitol and fructose as a result of hyperglycaemia, leading to deficiency of myoinositol.
5. Diabetic retinopathy. Diabetic retinopathy is a leading cause of blindness. There are 2 types of lesions involving retinal vessels: background and proliferative (page 508). Besides retinopathy, diabetes also predisposes the patients to early development of cataract and glaucoma.
6. Infections. Diabetics have enhanced susceptibility to various infections such as tuberculosis, pneumonias, pyelonephritis, otitis, carbuncles and diabetic ulcers. This could be due to various factors such as impaired leucocyte functions, reduced cellular immunity, poor blood supply due to vascular involvement and hyperglycaemia per se.
Diagnosis of Diabetes
Hyperglycaemia remains the fundamental basis for the diagnosis of diabetes mellitus. In symptomatic cases, the diagnosis is not a problem and can be confirmed by finding glucosuria and a random plasma glucose concentration above 200 mg/dl.
 The severity of clinical symptoms of polyuria and polydipsia is directly related to the degree of hyperglycaemia.
The severity of clinical symptoms of polyuria and polydipsia is directly related to the degree of hyperglycaemia.

 In asymptomatic cases, when there is persistently elevated fasting plasma glucose level, diagnosis again poses no difficulty.
In asymptomatic cases, when there is persistently elevated fasting plasma glucose level, diagnosis again poses no difficulty.
 The problem arises in asymptomatic patients who have normal fasting glucose level in the plasma but are suspected to have diabetes on other grounds and are thus subjected to oral glucose tolerance test (GTT). If abnormal GTT values are found, these subjects are said to have ‘chemical diabetes’ (Fig. 27.27). The American Diabetes Association (2007) has recommended definite diagnostic criteria for early diagnosis of diabetes mellitus (Table 27.8).
The problem arises in asymptomatic patients who have normal fasting glucose level in the plasma but are suspected to have diabetes on other grounds and are thus subjected to oral glucose tolerance test (GTT). If abnormal GTT values are found, these subjects are said to have ‘chemical diabetes’ (Fig. 27.27). The American Diabetes Association (2007) has recommended definite diagnostic criteria for early diagnosis of diabetes mellitus (Table 27.8).
The following investigations are helpful in establishing the diagnosis of diabetes mellitus:
I. URINE TESTING. Urine tests are cheap and convenient but the diagnosis of diabetes cannot be based on urine testing alone since there may be false-positives and false-negatives. They can be used in population screening surveys. Urine is tested for the presence of glucose and ketones.
1. Glucosuria. Benedict’s qualitative test detects any reducing substance in the urine and is not specific for glucose. More
TABLE 27.8: Revised Criteria for Diagnosis of Diabetes by Oral GTT (as per American Diabetes Association, 2007).
Plasma Glucose Value* |
Diagnosis |
|
|
FASTING (FOR > 8 HOURS) VALUE |
|
Below 100 mg/dl (< 5.6 mmol/L) |
Normal fasting value |
100-125 mg/dl (5.6-6.9 mmol/L) |
Impaired fasting glucose (IFG)** |
126 mg/dl (7.0 mmol/L) or more |
Diabetes mellitus |
TWO-HOUR AFTER 75 GM ORAL GLUCOSE LOAD |
|
< 140 mg/dl (< 7.8 mmol/L) |
Normal post-prandial GTT |
140-199 mg/dl (7.8-11.1 mmol/L) |
Impaired post-prandial glucose |
|
tolerance (IGT)** |
200 mg/dl (11.1 mmol/L) or more |
Diabetes mellitus |
RANDOM VALUE |
|
200 mg/dl (11.1 mmol/L) or more |
Diabetes mellitus |
in a symptomatic patient |
|
Note: * Plasma glucose values are 15% higher than whole blood glucose value.
** Individuals with IFG and IGT are at increased risk for development of type 2 DM later.
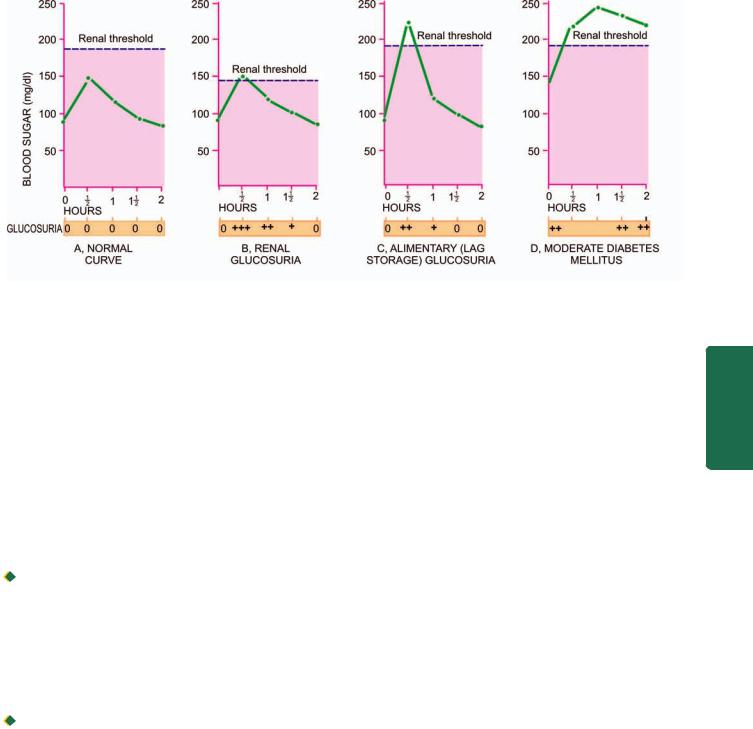
827
Figure 27.27 

 The glucose tolerance test, showing blood glucose curves (venous blood glucose) and glucosuria after 75 gm of oral glucose.
The glucose tolerance test, showing blood glucose curves (venous blood glucose) and glucosuria after 75 gm of oral glucose.
sensitive and glucose specific test is dipstick method based on enzyme-coated paper strip which turns purple when dipped in urine containing glucose.
The main disadvantage of relying on urinary glucose test alone is the individual variation in renal threshold. Thus, a diabetic patient may have a negative urinary glucose test and a nondiabetic individual with low renal threshold may have a positive urine test.
Besides diabetes mellitus, glucosuria may also occur in certain other conditions such as: renal glycosuria, alimentary (lag storage) glucosuria, many metabolic disorders, starvation and intracranial lesions (e.g. cerebral tumour, haemorrhage and head injury). However, two of these conditions—renal glucosuria and alimentary glucosuria, require further elaboration here.
 Renal glucosuria (Fig. 27.27,B): After diabetes, the next most common cause of glucosuria is the reduced renal threshold for glucose. In such cases although the blood glucose level is below 180 mg/dl (i.e. below normal renal threshold for glucose) but glucose still appears regularly and consistently in the urine due to lowered renal threshold.
Renal glucosuria (Fig. 27.27,B): After diabetes, the next most common cause of glucosuria is the reduced renal threshold for glucose. In such cases although the blood glucose level is below 180 mg/dl (i.e. below normal renal threshold for glucose) but glucose still appears regularly and consistently in the urine due to lowered renal threshold.
Renal glucosuria is a benign condition unrelated to diabetes and runs in families and may occur temporarily in pregnancy without symptoms of diabetes.
 Alimentary (lag storage) glucosuria (Fig. 27.27,C): A rapid and transitory rise in blood glucose level above the normal renal threshold may occur in some individuals after a meal. During this period, glucosuria is present. This type of response to meal is called ‘lag storage curve’ or more appropriately ‘alimentary glucosuria’. A characteristic feature is that unusually high blood glucose level returns to normal 2 hours after meal.
Alimentary (lag storage) glucosuria (Fig. 27.27,C): A rapid and transitory rise in blood glucose level above the normal renal threshold may occur in some individuals after a meal. During this period, glucosuria is present. This type of response to meal is called ‘lag storage curve’ or more appropriately ‘alimentary glucosuria’. A characteristic feature is that unusually high blood glucose level returns to normal 2 hours after meal.
2. Ketonuria. Tests for ketone bodies in the urine are required for assessing the severity of diabetes and not for diagnosis of diabetes. However, if both glucosuria and ketonuria are present, diagnosis of diabetes is almost certain. Rothera’s test (nitroprusside reaction) and strip test are conveniently performed for detection of ketonuria.
Besides uncontrolled diabetes, ketonuria may appear in individuals with prolonged vomitings, fasting state or exercising for long periods.
II. SINGLE BLOOD SUGAR ESTIMATION. For diagnosis of diabetes, blood sugar determinations are absolutely necessary. Folin-Wu method of measurement of all reducing substances in the blood including glucose is now obsolete. Currently used are O-toluidine, Somogyi-Nelson and glucose oxidase methods. Whole blood or plasma may be used but
whole blood values are 15% lower than plasma values.
A grossly elevated single determination of plasma glucose may be sufficient to make the diagnosis of diabetes.
A fasting plasma glucose value above 126 mg/dl (>7 mmol/L) is certainly indicative of diabetes. In other cases, oral GTT is performed.
III. SCREENING BY FASTING GLUCOSE TEST. Fasting plasma glucose determnitation is a screening test for DM type 2. It is recommended that all individuals above 45 years of age must undergo screening fasting glucose test every 3-years, and relatively earlier if the person is overweight or at risk because of the following reasons:
i)Many of the cases meeting the current criteria of DM are asymptomatic and donot know that they have the disorder.
ii)Studies have shown that type 2 DM may be present for about 10 years before symptomatic disease appears.
iii)About half the cases of type 2 DM have some diabetesrelated comlication at the time of diagnosis.
iv)The course of disease is favourably altered with treatment.
IV. ORAL GLUCOSE TOLERANCE TEST. Oral GTT is performed principally for patients with borderline fasting plasma glucose value (i.e. between 100-140 mg/dl). The patient who is scheduled for oral GTT is instructed to eat a high carbohydrate diet for at least 3 days prior to the test and come after an overnight fast on the day of the test (for at least 8 hours). A fasting blood sugar sample is first drawn.
System Endocrine The 27 CHAPTER

828 Then 75 gm of glucose dissolved in 300 ml of water is given. Blood and urine specimen are collected at half-hourly intervals for at least 2 hours. Blood or plasma glucose content is measured and urine is tested for glucosuria to determine the approximate renal threshold for glucose. Venous whole blood concentrations are 15% lower than plasma glucose values.
Currently accepted criteria for diagnosis of DM (as per American Diabetes Association, 2007) are given in Table 27.8:

 Normal cut off value for fasting blood glucose level is considered as 100 mg/dl.
Normal cut off value for fasting blood glucose level is considered as 100 mg/dl.
 Cases with fasting blood glucose value in range of 100125 mg/dl are considered as impaired fasting glucose tolerance (IGT); these cases are at increased risk of developing diabetes later and therefore kept under observation for repeating the test. During pregnancy, however, a case of IGT is treated as a diabetic.
Cases with fasting blood glucose value in range of 100125 mg/dl are considered as impaired fasting glucose tolerance (IGT); these cases are at increased risk of developing diabetes later and therefore kept under observation for repeating the test. During pregnancy, however, a case of IGT is treated as a diabetic.
|
Individuals with fasting value of plasma glucose higher |
|
|
than 126 mg/dl and 2-hour value after 75 gm oral glucose |
|
|
higher than 200 mg/dl are labelled as diabetics (Fig. 27.27,D). |
|
|
In symptomatic case, the random blood glucose value |
|
SECTION |
above 200 mg/dl is diagnosed as diabetes mellitus. |
|
V. OTHER TESTS. A few other tests are sometimes |
||
performed in specific conditions in diabetics and for research |
||
purposes: |
||
1. Glycosylated haemoglobin (HbA1C). Measurement of |
||
III |
blood glucose level in diabetics suffers from variation due |
|
to dietary intake of the previous day. Long-term objective |
||
|
||
Systemic |
assessment of degree of glycaemic control is better |
|
monitored by measurement of glycosylated haemoglobin |
||
(HbA1C), a minor haemoglobin component present in normal |
||
|
||
|
persons. This is because the non-enzymatic glycosylation of |
|
|
haemoglobin takes place over 90-120 days, lifespan of red |
|
Pathology |
blood cells. HbA1C assay, therefore, gives an estimate of |
|
Increased HbA1C value almost certainly means DM but |
||
|
diabetic control and compliance for the preceding 3-4 |
|
|
months. This assay has the advantage over traditional blood |
|
|
glucose test that no dietary preparation or fasting is required. |
|
|
normal value does not rule out IGT; thus the test is not used |
|
|
for making the diagnosis of DM. Moreover, since HbA1C |
|
|
assay has a direct relation between poor control and |
|
|
development of complications, it is also a good measure of |
|
|
prediction of microvascular complications. Care must be |
|
|
taken in iterpretation of the HbA1C value because it varies |
|
|
with the assay method used and is affected by presence of |
|
|
haemoglobinopathies, anaemia, reticulocytosis, transfusions |
|
|
and uraemia. |
|
|
2. Glycated albumin. This is used to monitor degree of |
|
|
hyperglycaemia during previous 1-2 weeks when HbA1C can |
|
|
not be used. |
|
|
3. Extended GTT. The oral GTT is extended to 3-4 hours for |
|
|
appearance of symptoms of hyperglycaemia. It is a useful |
|
|
test in cases of reactive hypoglycaemia of early diabetes. |
|
|
4. Intravenous GTT. This test is performed in persons who |
|
|
have intestinal malabsorption or in postgastrectomy cases. |
|
|
5. Cortisone-primed GTT. This provocative test is a useful |
|
|
investigative aid in cases of potential diabetics. |
6.Insulin assay. Plasma insulin can be measured by radioimmunoasay and ELISA technique. Plasma insulin deficiency is crucial for type 1 DM but is not essential for making the diagnosis of DM.
7.Proinsulin assay. Proinsulin is included in immunoassay of insulin; normally it is <20% of total insulin.
8.C-peptide assay. C-peptide is released in circulation during conversion of proinsulin to insulin in equimolar quantities to insulin; thus its levels correlate with insulin level in blood except in islet cell tumours and in obesity. This test is even more sensitive than insulin assay because its levels are not affected by insulin therapy.
9.Islet autoantibodies. Glutamic acid decarboxylase and islet cell cytoplasmic antibodies may be used as a marker for type 1 DM.
10.Screening for diabetes-associated complications.
Besides making the diagnosis of DM based on the defined criteria, screening tests are done for DM-associated complications e.g. microalbuniuria, dyslipidaemia, thyroid dysfunction etc.
ISLET CELL TUMOURS
Islet cell tumours are rare as compared with tumours of the exocrine pancreas. Islet cell tumours are generally small and may be hormonally inactive or may produce hyperfunction. They may be benign or malignant, single or multiple. They are named according to their histogenesis such as: β-cell tumour (insulinoma), G-cell tumour (gastrinoma), A-cell tumour (glucagonoma) D-cell tumour (somatostatinoma), vipoma (diarrhoeagenic tumour from D1 cells which elaborate VIP), pancreatic polypeptide (PP)-secreting tumour, and carcinoid tumour. However, except insulinoma and gastrinoma, all others are extremely rare and require no further comments.
Insulinoma (β-Cell Tumour)
Insulinomas or beta (β)-cell tumours are the most common islet cell tumours. The neoplastic β-cells secrete insulin into the blood stream which remains unaffected by normal regulatory mechanisms. This results in characteristic attacks of hypolgycaemia with blood glucose level falling to 50 mg/ dl or below, high plasma insulin level (hyperinsulinism) and high insulin-glucose ratio. The central nervous manifestations are conspicuous which are promptly relieved by intake of glucose. Besides insulinoma, however, there are other causes of hypoglycaemia such as: in starvation, partial gastrectomy, diffuse liver disease, hypopituitarism and hypofunction of adrenal cortex.
MORPHOLOGIC FEATURES. Grossly, insulinoma is usually solitary and well-encapsulated tumour which may vary in size from 0.5 to 10 cm. Rarely, they are multiple. Microscopically, the tumour is composed of cords and sheets of well-differentiated β-cells which do not differ from normal cells. Electron microscopy reveals typical crystalline rectangular granules in the neoplastic cells. It is extremely difficult to assess the degree of anaplasia to distinguish benign from malignant β-cell tumour.

Gastrinoma
(G-Cell Tumour, Zollinger-Ellison Syndrome)
Zollinger and Ellison described diagnostic triad consisting of the following:

 Fulminant peptic ulcer disease
Fulminant peptic ulcer disease

 Gastric acid hypersecretion
Gastric acid hypersecretion

 Presence of non-β pancreatic islet cell tumour.
Presence of non-β pancreatic islet cell tumour.
Such non-β pancreatic islet cell tumour is the source of gastrin, producing hypergastrinaemia and hence named gastrinoma. Definite G cells similar to intestinal and gastric G cells which are normally the source of gastrin in the body, have not been identified in the normal human pancreas but neoplastic cells of certain islet cell tumours have ultrastructural similarities.
MORPHOLOGIC FEATURES. Majority of gastrinomas occur in the wall of the duodenum. They may be benign or malignant. Gastrinomas are associated with peptic ulcers at usual sites such as the stomach, first and second part of the duodenum, or sometimes at unusual sites such as in the oesophagus and jejunum. About one-third of patients have multiple endocrine neoplasia—multiple adenomas of the islet cells, pituitary, adrenal and parathyroid glands.
MISCELLANEOUS ENDOCRINE TUMOURS
MULTIPLE ENDOCRINE NEOPLASIA (MEN) SYNDROMES
Multiple adenomas and hyperplasias of different endocrine organs are a group of genetic diorders which produce heterogeneous clinical features called multiple endocrine neoplasia (MEN) syndromes. Presently, 4 distinct types of MEN syndromes are distinguished. These are briefly outlined below along with major disease associations:
1. MEN type 1 syndrome (Wermer’s syndrome) includes adenomas of the parathyroid glands, pancreatic islets and pituitary. The syndrome is inherited as an autosomal dominant trait. There is 50% chance of transmitting the predisposing gene, MEN 1 (or menin) gene, to the child of an affected person. MEN 1 is characterised by the following features:
1. Parathyroid: Hyperplasia or adenoma; hyperparathyroidism is the most common (90%) clinical manifestation.
2.Pancreatic islet cells: Hyperplasia or adenoma seen in 80% cases; frequently with Zollinger-Ellison syndrome.
3.Pituitary: Hyperplasia or adenoma in 65% cases; manifest as acromegaly or hypopituitarism.
4.Adrenal cortex: Uncommonly involved by adenoma or pheochromocytoma.
5.Thyroid: Less commonly involved by adenoma or hyperplasia.
2. MEN type 2 syndrome (Sipple’s syndrome) is characterised by medullary carcinoma thyroid and pheochromocytoma. Genetic abnormality in these cases is mutation in RET gene in almost all cases. MEN 2 has two major syndromes:

 MEN type 2A is the combination of medullary carcinoma thyroid, pheochromocytoma and hyperparathytroidism. MEN type 2A has further three subvariants:
MEN type 2A is the combination of medullary carcinoma thyroid, pheochromocytoma and hyperparathytroidism. MEN type 2A has further three subvariants:
i)MEN 2A with familial medullary carcinoma thyroid
ii)MEN 2A with cutaneous lichen amyloidosis
iii)MEN 2A with Hirschsprung’s disease.
 MEN type 2B the combination of medullary carcinoma thyroid, pheochromocytoma, mucosal neuromas, intestinal ganglioneuromatosis, and marfanoid features.
MEN type 2B the combination of medullary carcinoma thyroid, pheochromocytoma, mucosal neuromas, intestinal ganglioneuromatosis, and marfanoid features.
3. Mixed syndromes include a variety of endocrine neoplastic combinations which are distinct from those in MEN type 1 and type 2. A few examples are as under:
 von Hippel-Lindau syndrome from mutation in VHL gene is association of CNS tumours, renal cell carcinoma, pheochromocytoma and islet cell tumours.
von Hippel-Lindau syndrome from mutation in VHL gene is association of CNS tumours, renal cell carcinoma, pheochromocytoma and islet cell tumours.

 Type 1 neurofibromatosis from inactivation of neurofibromin protein and activation of RAS gene, is associated with MEN type 1 or type 2 features.
Type 1 neurofibromatosis from inactivation of neurofibromin protein and activation of RAS gene, is associated with MEN type 1 or type 2 features.
POLYGLANDULAR AUTOIMMUNE (PGA) SYNDROMES
Immunologic syndromes affecting two or more endocrine glands and some non-endocrine immune disturbances produce syndromic presentation termed polyglandular autoimmune (PGA) syndromes. PGA syndromes are of two types:
 PGA type I occurring in children is characterised by mucocutaneous candidiasis, hypoparathyroidism, and adrenal insufficiency.
PGA type I occurring in children is characterised by mucocutaneous candidiasis, hypoparathyroidism, and adrenal insufficiency.

 PGA type II (Schmidt syndrome) presents in adults and commonly comprises of adrenal insufficiency, autoimmune thyroiditis, and type 1 diabetes mellitus.
PGA type II (Schmidt syndrome) presents in adults and commonly comprises of adrenal insufficiency, autoimmune thyroiditis, and type 1 diabetes mellitus.
829
System Endocrine The 27 CHAPTER