

спектроскопические методы анализа
.pdfуровню соответствует набор колебательных уровней. Поэтому один и тот же электронный переход может происходить с различных колебательных уровней основного состояния и заселять несколько колебательных уровней, принадлежащих данному возбужденному электронному состоянию. Таким образом, электронные переходы в действительности соответствуют комбинированным, электронно-колебательным переходам, что определяет вид кривой спектра поглощения.
3. Энергия фотонов видимого света 400 – 760 нм, с поглощением которых связана окраска вещества, составляет всего лишь 300 – 160 кДж/моль. Такого количества энергии достаточно для возбуждения электронов только наружных слоев, т. е. валентных электронов довольно сложных молекул на ближайшие свободные энергетические уровни. В связи с этим важнейшим условием для понимания причин появления окраски станет выяснение зависимостей между структурой молекулы и распределением в ней электронных уровней.
Конфигурация электронно-колебательных уровней
Образование ковалентной связи между двумя атомами X и Y (рис. 3.16) сопровождается:
-повышением электронной плотности в области между ядрами;
-сближением атомов с их фиксацией на определенном расстоянии r0, которое соответствует длине этой связи;
-понижением энергии молекулярной системы на величину, равную энергии образовавшейся связи.
r0
X


 Y
Y
Eсвязи
Рис. 3.16. Образование ковалентной связи между двумя атомами X и Y
31
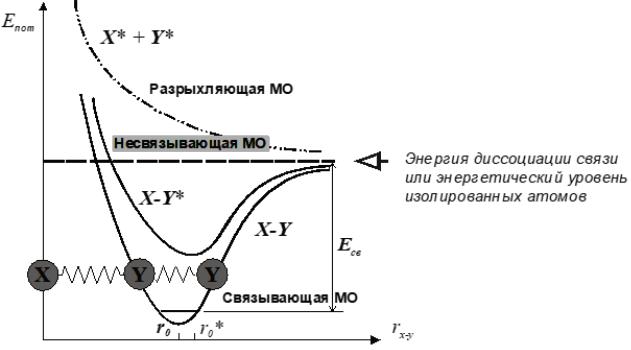
Таким образом, электронная энергия связи для двухатомной
молекулы является функцией межъядерного расстояния: |
|
Е пот = f (rx-y) |
(10) |
Графически такую зависимость можно представить в |
виде |
характеристической кривой, имеющей форму искаженного параболоида, с энергетическим минимумом над точкой равновесного межъядерного расстояния r0.
Эта энергетическая кривая соответствует связывающей молекулярной орбитали (рис. 3.17).
Рис. 3.17. Зависимость энергии связи от межъядерного расстояния
Минимум на кривой возникает потому, что при дальнейшем сближении атомов начинают преобладать силы межъядерного отталкивания. Расхождение атомов на большее расстояние также требует дополнительной энергии для преодоления сил ковалентного связывания. По мере увеличения межъядерного расстояния правая ветвь кривой приближается к энергетическому уровню изолированных атомов и, наконец, наступает диссоциация молекулы. Таким образом, разность между уровнем энергии диссоциированных атомов и нулевым уровнем
32
молекулы соответствует энергии данной связи Есв или равна энергии диссоциации Едис, взятой с обратным знаком.
Если сближение атомов не сопровождается возникновением связи между ними, характер энергетической кривой существенно изменяется. По мере уменьшения межъядерного расстояния rx-y силы отталкивания быстро нарастают, и энергетическая кривая приобретает вид экспоненты, что соответствует разрыхляющей молекулярной орбитали.
В возбужденном состоянии электрон заселяет более высоко расположенный вакантный электронный уровень и осуществляет одноэлектронную связь в молекуле. Энергия такой связи значительно меньше обычной двухэлектронной связи, поэтому длина связи r0 несколько увеличивается. При этом соответствующая кривая зависимости энергии от межъядерного расстояния, хотя и сохраняет форму параболоида, потенциальная яма становится менее глубокой и ее правый склон более пологим. Понятно, что для высших возбужденных электронных состояний эта тенденция еще больше усиливается и, в конце концов, межатомная связь практически исчезает, а параболическая кривая превращается в экспоненту.
Для многоатомной молекулы зависимость Епот от межъядерных расстояний превращается в многомерную гиперповерхность, которая имеет энергетические минимумы, соответствующие равновесным конфигурациям атомов. Гиперповерхность описывается в многопараметровой системе координат. Такая модель сложна и не обладает наглядностью. Поэтому приближенно используют сечение потенциальной гиперповерхности по координате наиболее слабой в данной молекуле связи. Эта координата сильнее других изменяется в ходе фотофизических процессов. Таким образом, многомерный случай сводят к двухмерному, который мы рассмотрим более подробно.
Спектры поглощения органических соединений состоят из широких полос, а не узких линий. Это объясняется тем, что любая молекула при фотовозбуждении изменяет не только электронную, но и колебательную энергию.
Колебания связанных ядер вдоль межъядерной оси можно рассматривать как колебания ядра Y относительно неподвижного ядра X.
33

При небольших отклонениях ядра Y от положения равновесия ro молекулу приближенно рассматривают как гармонический осциллятор, а зависимость величины потенциальной энергии Епот от отклонения ядер изображается искаженным параболоидом (так называемая кривая Морзе). Общая колебательная энергия ядер – сложная величина:
Екол = Епот + Екин |
(11) |
Из-за относительно малой массы ядер их колебания квантуются. |
|
Для данного электронного состояния |
колебательная энергия |
принимает лишь определенные величины в соответствии со значениями колебательных квантовых чисел ν = 0, 1, 2, 3 и т.д. и может быть вычислена по уравнению Морзе:
E |
(ν) = hν |
(ν + 1⁄ |
) − |
h2ν2 |
(ν+1⁄ |
)2 |
|
(12) |
кол |
2 |
, |
||||||
|
|
|
|
|
|
|
|
|
кол |
кол |
2 |
|
|
4D |
|
|
|
где ν – колебательное квантовое число; νкол – частота колебания ядер;
D – энергия диссоциации связи.
Из этого уравнения следует, что низшая колебательная энергия – энергия нулевого колебательного уровня – отлична от нуля, так как Eкол(0)
1/2 hν.
Теперь рассмотрим некоторые свойства потенциальной кривой электронного состояния колебательной системы, рис. 3.18.
Рис. 3.18. Потенциальная кривая электронного состояния и квантование энергии колебания ядер (функция Морзе)
34
Точки пересечения разрешенных колебательных уровней полной энергии с потенциальной кривой (например, точки А и В) можно сопоставить с точками максимальной амплитуды колебаний гармонического осциллятора. В этих точках вся кинетическая энергия осциллятора превращается в потенциальную энергию. В промежуточных точках горизонтальных отрезков кинетическая энергия колеблющихся атомов отличается от нуля. Согласно уравнению, при возрастании колебательного квантового числа колебательные уровни сближаются.
В виде синусоидальных кривых на рисунке показано распределение плотности вероятности нахождения ядра Y на определенном межъядерном расстоянии. При повышении колебательной энергии возрастает вероятность нахождения ядра вблизи потенциальной кривой. Напротив, на нулевом колебательном уровне максимум вероятности нахождения ядра Y совпадает с равновесным межъядерным расстоянием r0.
Форма потенциальной кривой особенно сильно отличается от симметричной параболы в области малых межъядерных расстояний. Вследствие эффекта отталкивания ядер она круто идет вверх. В области больших межъядерных расстояний из-за ослабления ковалентной связи кривая идет более полого. Поэтому энергетику потенциальной кривой описывают функцией Морзе, которая при определении колебательной энергии учитывает энергию диссоциации связи.
Принцип Франка – Кондона и форма полос поглощения
Конфигурация полос поглощения в электронном спектре существенно зависит от избыточной колебательной энергии, которую молекула получает при электронном возбуждении. Приращение колебательной энергии непосредственно связано:
–с различием межъядерных расстояний в основном и возбужденном состояниях молекулы;
–изменением формы соответствующих потенциальных кривых. Происхождение этой избыточной колебательной энергии
устанавливает принцип Франка – Кондона.
35
Он гласит: электронный переход происходит за гораздо меньшее время (10 15 – 10 14 с), чем период колебаний атомных ядер (10 12 – 10 10 с), поэтому за время электронного возбуждения относительное расположение атомов (геометрия молекул) практически не изменяется.
Если процессы фотовозбуждения изображать как переход с одной потенциальной кривой на другую, то принцип Франка – Кондона приводит
кследующим трем правилам.
1.Электронно-колебательные переходы происходят вертикально при неизменных межъядерных расстояниях.
2.Наиболее вероятны и интенсивны переходы с колебательных уровней основного электронного состояния, которые начинаются в области максимальной вероятности нахождения колеблющегося ядра. Для нулевого колебательного уровня (v=0) это расположение ядер вблизи равновесного состояния r0. Для более высоких колебательных уровней (v=1,2,3,…) – вблизи потенциальной кривой. Следует помнить, что при комнатной температуре только часть молекул может находиться на низшем колебательном уровне.
3.При прочих равных условиях наиболее вероятны и интенсивны такие переходы, которые заканчиваются в области максимальной вероятности нахождения ядер на соответствующем колебательном уровне возбужденного состояния. Поэтому электронный переход обычно заканчивается вблизи потенциальной кривой возбужденного состояния (левой его ветви).
Рассмотрим два крайних случая формирования полосы поглощения в электронном спектре.
Если равновесные межъядерные расстояния в основном и возбужденном состояниях практически одинаковы и минимум верхней кривой находится над минимумом нижней кривой (основное состояние), то полоса электронного перехода в спектре поглощения оказывается узкой, асимметричной, с колебательной структурой (рис. 3.19).
36

Рис. 3.19. Форма полосы поглощения в электронном спектре в случае одинаковых межъядерных расстояний в основном и возбужденном состояниях молекулы
Вэтом случае наиболее интенсивным является 0 – 0 переход как наиболее вероятный. Он проявляется в спектре как самый длинноволновый из нескольких колебательных максимумов 0 – 1, 0 – 2, 0 – 3 меньшей интенсивности.
Однако при электронном возбуждении связи в молекуле, как правило, ослабляются. Минимум потенциальной кривой возбужденного состояния соответствует большему межъядерному расстоянию, чем в основном состоянии (рис. 3.20).
Вэтом случае наиболее интенсивный колебательный максимум уже не соответствует 0 – 0 переходу, поскольку теперь он менее вероятен. Более интенсивными и более вероятными оказываются переходы на следующие колебательные уровни 0 – 2 или 0 – 3.
Колебательные максимумы уширяются, и в результате их перекрывания в спектре наблюдается широкая и симметричная полоса электронного перехода. При записи спектра в полярном растворителе тонкая структура полосы поглощения часто совсем не обнаруживается. Обычно это следствие лучшей сольватации молекул, возбужденных светом.
Из сказанного следует, что ширину полосы можно рассматривать как меру изменения межъядерных расстояний при электронном возбуждении.
37
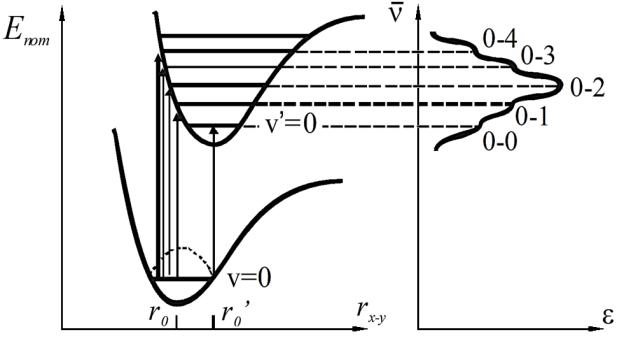
Рис. 3.20. Форма полосы поглощения в электронном спектре в случае различных межъядерных расстояний в основном и возбужденном состояниях молекулы
Итак, один и тот же электронный переход в молекуле совершается при поглощении молекулой фотонов различных, но близких энергий.
Результирующая спектральная полоса состоит из большого числа линий электронно-колебательных переходов, которые, перекрываясь, образуют широкую полосу поглощения. Так как вероятность комбинированных переходов различна, полоса приобретает максимум и склоны различной крутизны.
Следственно, значение макс полосы в электронном спектре является мерой энергии электронного возбуждения молекулы. А величина макс служит мерой вероятности того, что при поглощении фотона данной энергии молекула перейдет в возбужденное состояние.
Энергии электронных состояний зависят от природы и строения молекул, а значит, электронные спектры поглощения могут дать ценную информацию об их свойствах. Приближенно можно считать, что изменение электронного состояния молекулы в результате поглощения одного фотона (кванта) излучения достигается за счет перехода одного из электронов в электронной оболочке молекулы на одну из вышележащих пустых молекулярных орбиталей. Из условия Бора следует, что чем
38
меньше разница в энергиях молекулярных орбиталей, между которыми осуществляется электронный переход, тем больше длина волны излучения, требуемая для этого перехода.
Согласно положениям теории молекулярных орбиталей для двухатомных молекул, линейная комбинация двух атомных орбиталей (АО) взаимодействующих атомов приводит к возникновению двух молекулярных орбиталей (МО), одна из которых энергетически более выгодна, чем любая из исходных АО, а вторая – энергетическим менее выгодна, чем любая из АО, участвующих в комбинации. Первая из этих МО называется связывающей, а вторая – разрыхляющей. Разница в энергиях между связывающей и разрыхляющей орбиталями зависит от типа взаимодействия между участвующими АО: чем больше область перекрывания соответствующих волновых функций, тем больше эта разница. Пользуясь общепринятыми условными изображениями атомных орбиталей, можно оценить величину этой области в зависимости от типа перекрывания. Очевидно, что при заданном межатомном расстоянии лобовое перекрывание (δ-тип) будет более эффективным, чем боковое (π- тип). Следовательно, разница в энергиях между δ-связывающей и δ*- разрыхляющей МО будет существенно большей, чем между π- связывающей и π*-разрыхляющей МО (рис. 3.21).
Отсюда следует, что молекулы, не имеющие π-орбиталей (т. е. кратных связей), могут иметь полосы поглощения, соответствующие δ→δ* переходам, требующим высокой энергии, а потому наблюдаемым в далекой УФ-области, недоступной для стандартных приборов. Полос поглощения в ближней УФ-области следует ожидать от переходов типа π→π*, требующих меньшей энергии, а потому характеризуемых большей длиной волны.
Если у одного из атомов имеются АО, которые не могут дать эффективного перекрывания ни с одной из АО другого атома, то они останутся неизменными по своему виду и энергии, и будут считаться несвязывающими, или n-орбиталями. Они бывают заполнены парами
39

электронов, которые называют неподеленными электронными парами. Поскольку n-орбитали являются, по сути дела, атомными орбиталями, то им соответствует более высокая энергия, чем энергия связывающих МО. В молекулах, не содержащих π-орбиталей, однако имеющих заполненные электронами n-орбитали, могут осуществляться n→δ* переходы, но положение соответствующих им полос в спектре будет сильно зависеть от природы атома, несущего неподеленную пару электронов.
Рис. 3.21. Общая схема электронных переходов
Основные типы электронных переходов в органических молекулах и их характеристики представлены в табл. 3.4.
40