

5 quants / Лабораторный практикум Квантово-химическое моделирование соединений в пакете HyperChem Учебно-методическое пособие
.pdf•Когда нет возможности провести оптимизации (очень большие системы, более 1000 электронов).
•Если имеется экспериментальная информация о характеристиках конфигурации.
8.3. Расчет в одной точке c учетом конфигурационного взаимодействия (Single Point CI)
Команда Single Point CI (Configuration Interaction - Конфигура-
ционное взаимодействие) используется для активации расчета конфигурационных взаимодействий и открывает соответствующее диалоговое окно (рис. 1.53). Такой подход необходимо применять при расчетах УФ и оптических спектров в видимом диапазоне, а также возбужденных состояний. Выбор этой опции существенно увеличивает время расчетов, так что проводится в фиксированной предварительно оптимизированной геометрии.
8.3.1. Диалоговое окно параметров конфигурационного взаимодействия
Учет конфигурационного взаимодействия может быть использован для улучшения качества волновой функции и энергии состояния. Все расчеты в приближении самосогласованного поля (SCF) основаны на одноэлектронной модели, суть которой заключается в том, что каждый электрон движется в усредненном поле, которое формируется всеми остальными электронами. Считается, что электроны взаимодействуют мгновенно и стремятся избегать друг друга согласно принципу Паули. Такая корреляция приводит к понижению среднего межэлектронного отталкивания и, в свою очередь, к понижению энергии состояния. Отличие между полной энергией, рассчитанной в SCF подходе, и точной энергией, полученной в нерелятивистском подходе, называется корреляционной энергией.
КВ (CI) расчеты, возможно, являются наиболее широко распространенным методом выхода за пределы SCF-подхода. Результатом SCF расчета является конфигурация состояния, в котором одноэлектронные уровни жестко заполнены электронами. Другие конфигурации могут быть сформированы из конфигурации, полученной в самосогласованном расчете при помощи возбуждения электронов из занятых на виртуальные (вакантные) орбитали. Результатом КВ расчета является набор улучшенных состояний, каждое из
61
которых представляется линейной комбинацией таких конфигураций. КВ расчеты невозможно проводить в режиме оптимизации геометрии, а можно только в одной точке (single point). В методе РМХ этот подход также не реализован.
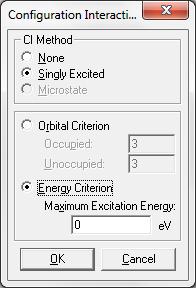
Для установки параметров КВ расчетов используется диалоговое окно Configuration Interaction (Конфигурационное взаимодействие) (рис. 1.53). Вначале необходимо выбрать соответствующий полуэмпирический или неэмпирический метод расчета. Вам доступны следующие параметры:
Рис. 1.53. Опции метода конфигурационного взаимодействия
None (ни одного) – расчет конфигурационных взаимодействий производиться не будет.
Singly Exited (однократно возбужденное) – в расчете будут учитываться только однократно возбужденные состояния.
Microstate (микросостояние) – означает, что в расчете кроме од- нократно-возбужденных состояний будут учитываться и все возможные многократные.
Orbital Criterion (орбитальный критерий) – выбор этого параметра определяет диапазон орбиталей, с которых и на которые происходят электронные возбуждения, формирующие взаимодействующие конфигурации.
• Occupied (занятые) – определяет область занятых орбиталей, начиная с высшей занятой молекулярной орбитали (ВЗМО – HOMO), с которой происходит возбуждение орбиталей.
62
•Unoccupied (вакантные) – определяет область вакантных (виртуальных) орбиталей, начиная с низшей вакантной орбитали (НВМО), участвующих в электронных возбуждениях.
Energy Criterion (энергетический критерий) – является опцией орбитального критерия (высшее значение энергии МО), который устанавливает ограничения по энергии при генерировании набора взаимодействующих конфигураций. Эта опция доступна только для однократно возбужденных конфигураций (Singly Exited).
•Maximum Exitation (максимальное возбуждение) – определяет наибольшую разницу по энергии (в эВ) между занятыми и вакантными орбиталями, включенными в CI расчет. В общем виде, конфигурации с высокой энергией не могут сильно взаимодействовать с конфигурацией основного состояния. Чем выше этот параметр, тем больше конфигураций включается в CI расчет.
8.3.2. Назначение метода конфигурационного взаимодействия
Практическое применение расчетов методом конфигурационного взаимодействия можно использовать при вычислении:
•УФ и оптических спектров;
•энергии возбужденных состояний;
•изучения создания или разрыва химических связей, изменения спинового состояния;
•описания вырожденных или близких к вырождению состояний;
•изучения расщепления синглет-триплет на более высоком
уровне.
Метод микросостояний понижает энергию некоррелированного и возбужденных состояний. Метод однократного возбуждения электронов предназначен только для расчетов УФ и видимых спектров
ине улучшает энергию основного состояния. Необходимо внимательно использовать энергетический критерий. Этот критерий должен быть больше, нежели энергетическая щель между занятыми
ивакантными орбиталями. В больших системах, как правило, в небольших энергетических интервалах находится большое количество орбиталей. Следовательно, размер CI матрицы может быть очень чувствительным к величине энергетического критерия, а время вычислений сильно зависит от размера CI матрицы и, следовательно, необходим вычислительный ресурс.
63
II. ПРАКТИЧЕСКИЕ ПРИМЕРЫ РАСЧЕТА МОЛЕКУЛЯРНЫХ ХАРАКТЕРИСТИК
Имеющиеся методы квантовой химии в программе HyperChem позволяют рассчитать большой набор свойств молекулярных структур. Наиболее важными при решении частных задач вычислительной химии являются энергетические характеристики, геометрия, спектр энергий электронов и форма молекулярных орбиталей, заряды атомов, дипольный момент и электростатический потенциал. Естественно, что сравнение свойств различных молекул возможно только в случае использования для расчета одного и того же выбранного метода.
9. Минимизация энергии системы и поиск стабильной конформации
В этом упражнении мы выполним:
•одноточечные вычисления и оптимизацию геометрии методом молекулярной механикой
•отражение фрагмента молекулы через определенную плоскость
•использование торсионных связей для контроля состояния и структурных свойств системы С этого упражнения мы начнем использовать инструменты ана-
лиза HyperChem, которые помогут нам понять поведение молекул и их взаимодействие. В качестве первого объекта, энергию которого
мы будем минимизировать, выступит циклогексан C6H12. Прежде, чем начать, важно понять некоторых из основных концепций минимизации энергии.
Минимизация энергии изменяет геометрию молекул для понижения энергии системы и в результате находится более устойчивая конформация. Во время минимизации, программа ищет молекулярное строение, в котором энергия не изменяется с бесконечно малыми изменениями в геометрии. Это означает, что производная энергии относительно всех координат, называемых градиентом, равна нулю. Она известна как стационарная точка на поверхности потенциальной энергии. Если незначительные изменения в геометрических параметрах увеличивают энергию молекулы, а конформация относительно устойчива, то она будет называться минимумом
64
энергии. Если энергия при незначительных изменениях координат понижается в одном или нескольких измерениях, но не во всех, это
- седловая точка.
Молекулярная система может иметь много минимумов энергии. Система с самой низкой энергией называется - глобальный минимум, а остальные будут называться - локальные минимумы.
В этом упражнении мы разберем три стационарные точки минимума энергии для циклогексана: «кресло», «ванна», и «твистванна». Также их называют конформерами. Для каждого конформера мы выполним оптимизацию методом молекулярной механики и сравним энергии, чтобы определить конформацию с глобальной минимальной энергией.
Удобнее всего регистрацию результатов вычислений получать не из строки состояния, а из log-файла, где сохраняется и другая информация. Запуск файла регистрации выполняется в меню File > Start Log. Файлу дают название и устанавливают степень полноты записи протокола молекулярно-механического «Mechanics Print Level» = 9 и квантово-химического расчета «Quantum print level» = 9. Если файл log уже существует, Вы можете добавлять к нему информацию, используя к окне опцию Append.
9.1. Выбор силового поля молекулярной механики
Прежде, чем Вы построите из циклогексана структуру «кресла» и выполнить оптимизацию молекулярной механикой, Вы должны выбрать силовое поле молекулярной механики, включенное в HyperChem. Силовое поле содержит типы атомов и параметры, которые должны быть назначены на молекулу прежде, чем Вы выполните вычисление молекулярной механикой. Для этого упражнения, мы будем использовать силовое поле AMBER.
Настройка силового поля:
1.Выбрать Molecular Mechanics на меню Setup.
2.В диалоге выбрать AMBER.
3.L-щелчок на Options, чтобы открыть ячейку диалога Force Field Options.
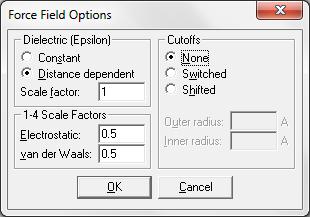
4.Установить параметр Distance Dependent на Dielectric.
Это необходимо, если при вычислении не используются молекулы растворителя, типа воды, а зависимую от расстояния диэлектрическую проницаемость, в которой Кулоновские взаимодействия
65
уменьшаются по закону ~ 1/r2, т.е. быстрее, чем по закону ~ 1/r при участии растворителя.
5.Установить Scale factor на 1.
6.Выбрать и Electrostatic и van der Waals 1-4 коэффициенты масштаба в 0.5 (рис. 2.1).
Эти выборы определяют масштабирование несвязанных взаимодействий для атомов, которые отделены тремя связями.
7.Выберите Cutoffs как None.
Для вычислений на больших структурах, желательно уменьшить диапазон вычислений, игнорируя взаимодействия дальнего порядка. В случае, когда структура достаточно маленькая, этот выбор может игнорироваться.
8. Нажать OK для закрытия обеих диалоговых окон.
Рис. 2.1. Параметры метода Amber
Для силового поля AMBER доступны несколько различных наборов параметров, и пользователи могут определять собственные наборы параметров. Выберите Select Parameter Set в меню Setup и
вокне диалога, выберите amber2.
9.2.Строительства циклогексана в форме конформации «кресло»
1. Установить в Default Element углерод, и выбрать Draw. 2. Установить в меню Select опцию Atoms.
3. В меню Display > Labels маркировать атомы номерами.
4. Удостоверитесь, что в меню Build выключено Explicit Hydrogens.
5. Нарисовать 2-D структуру, как показано на рис. 2.2.
66
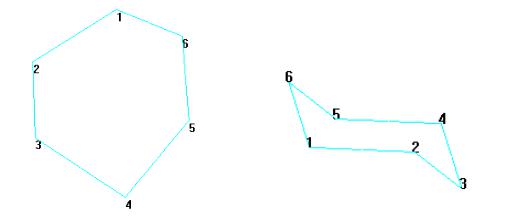
6.Выбрать Add H & Model Build в меню Build.
7.Выключить Show Hydrogens в меню Display.
8.Вращать и перемещать структуру, пока она не будет напоминать правую часть рис. 2.2.
Данная структура не оптимизирована, но она имеет стандартный набор длин связи, углов и торсионных скручиваний.
Измерение структуры циклогексана в форме «кресла»
Измерим структурные свойства сформированной неоптимизированной модели для дальнейшего сравнения с параметрами геометрии оптимизированной структуры.
|
Рис. 2.2. 2D эскиз и конформация «кресло» циклогексана |
||
Измерение геометрии молекулы |
|||
1. |
Выбрать инструмент Select. |
||
2. |
Установить метку на Atoms и снять метку с Multiple Selection. |
||
3. |
Выбрать несколько связей С-С, валентных и торcионных углов |
||
для исследования геометрии структуры. |
|||
Длина связи С-С: |
1,54Å |
|
|
Валентный угол: |
109,47 град. |
|
|
Торсионный угол: |
60 град |
рабочего пространства отмените |
|
4. |
R-щелчком в |
пустой области |
|
выделение.
Выполнение расчета в одной точке (single point)
Проведем одноточечное вычисление, чтобы получить полную энергию не оптимизированной конфигурации. Выберите Single Point в меню Compute.
Одноточечное вычисление сообщает нам в строке состояния об
67
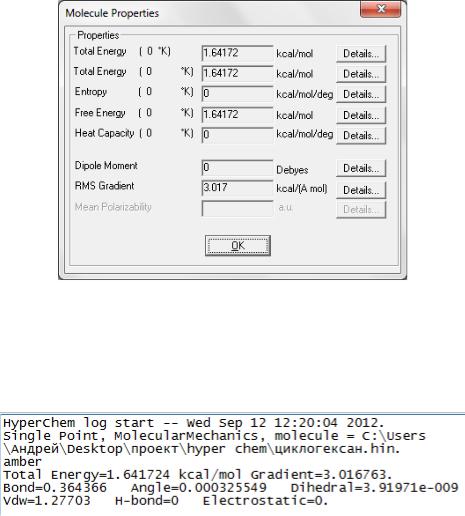
энергии в ккал/моль и общем среднеквадратичном (RMS) градиенте в kcal/(моль Ангстрем) текущей конфигурации атомов:
Энергия |
1,64 kcal/mol |
Градиент |
3,02 kcal/(A mol) |
В локальном минимуме, градиент должен быть почти равен нулю. Таким образом, сформированная модельная структура - не соответствует локальному минимуму, используя для вычислений силовое поле AMBER.
В качестве полезного замечания покажем, что энергетические характеристики после расчета можно получить несколькими альтернативными способами. Во-первых, через меню Compute > Properties (рис. 2.3).
Рис. 2.3. Энергия молекулы
Во-вторых, вся информация одноточечного вычисления сохраняется в log-файле (рис. 2.4). Файл регистрации также показывает и другие компоненты энергии.
Рис. 2.4. Фрагмент log-файла расчета методом amber
9.3. Оптимизация геометрии структуры «кресло»
Следующим шагом мы должны минимизировать энергию структуры «кресла», выполняя оптимизацию методом молекулярной ме-
68
ханики. Сначала необходимо установить параметры минимизации, включая алгоритм минимизации.
Установка параметров геометрической оптимизации
1.Выбрать Geometry Optimization в меню Compute.
2.Выберите для RMS градиента - 0.1.
L-щелчок на OK приведет к старту вычисления. С началом оптимизация геометрии начинается движение молекулы и смена информации в строке состояния. По окончании расчета в строке состояния появится:
Энергия |
1.33 kcal/mol |
Градиент |
0.07 kcal/(A mol) |
Градиент энергии стал намного меньше, чем градиент, измеренный для сформированной модельной структуры.
Измерение геометрии минимизированной системы
Теперь сравните структурные свойства (связи, валентные и торсионные углы) минимизированной системы с таковыми для сформированной модельной структуры.
|
Без оптимизации |
С оптимизацией |
Длина связи: |
1.54Å |
1.53Å |
Валентный угол: |
109.47 град. |
110.2 град |
Торсионный угол: |
60 град |
58.0 град |
Сравнение свойств структуры показывает, что минимизация энергии увеличила немного тетраэдрический угол и длину связи и уменьшила торсионный угол на 2 градуса.
9.4.Преобразование циклогексана из конформации «кресло»
в«ванну»
Чтобы вывести форму «ванны» для циклогексана необходимо зеркально отразить один конец молекулы.
Определение отражательной плоскости:
1.Пометить Multiple Selections в меню Select.
2.Выберите инструмент Select.
3.Сделать двойной щелчок на инструменте Select, чтобы возвратиться сформированной структурной модели.
69
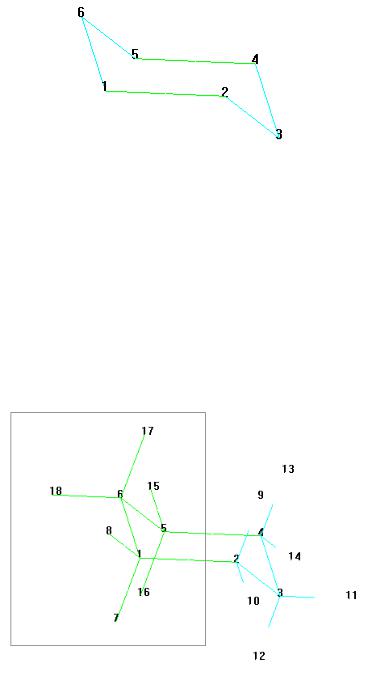
4. Щелкните на связях атомов углерода 1-2 и 5-4, чтобы выбрать отражательную плоскость, как показано на рис. 2.5.
Рис. 2.5. Выбор отражательной плоскости
5.Выбрать Name Selection в меню Select.
6.Выбрать PLANE (of Selection), и затем OK.
Отражение одного конец молекулы
1. Выберите Show Hydrogens и используйте Zoom, чтобы масштабировать молекулу так, чтобы вся молекула была видна. 2.Сделать LR-протяжку мышью, чтобы включить в выбор все атомы по одну сторону молекулы (рис .2.6).
3. Выбрать Reflect в меню Edit.
Рис. 2.6. Выбор группы атомов для отражения
Выбранные атомы отражаются через плоскость PLANE, производя преобразование формы циклогексана в новую структуру
«ванна» (рис. 2.7).
4. Щелкните в пустой области рабочего пространства правой кнопкой для снятия выделения атомов.
70