

IV.3.2.2. Реакции электрофильного замещения в ароматическом ядре фенолов.
Группа ОН фенолов, обладающая значительным +М-эффектом, является сильным донором; еще более сильным донором (сильнейшим из всех обычно встречающихся заместителей) является отрицательно заряженный атом кислорода в фенолят-ионе, обладающий как +М, так и +I-эффектами. Поэтому реакции электрофильного замещения в ароматическом ядре фенолов и особенно фенолятов идут чрезвычайно легко. При этом здесь протекают как «классические» реакции электрофильного замещения, характерные для аренов, так и другие электрофильные реакции, которые с аренами идти не могут.
А. Рассмотренные ранее реакции «классического» ароматического электрофильного замещения (нитрование, сульфирование, галогенирование в ядро, алкилирование и ацилирование по Фриделю-Крафтсу) идут для фенолов гораздо легче, чем для аренов. Здесь в целом ряде случаев можно использовать гораздо более мягкие реагенты и условия.
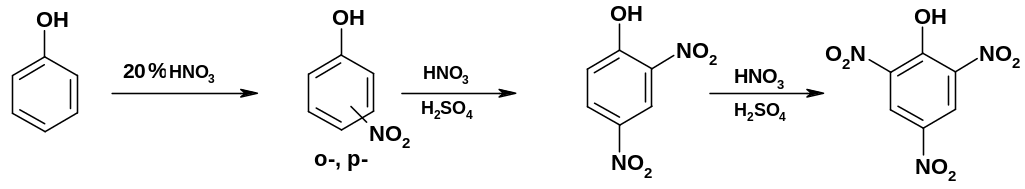
1. При нитровании фенола разбавленной азотной кислотой образуются о- и р-нитрофенолы; при действии на последние конц. HNO3 или нитрующей смеси образуются 2,4-динитрофенол и далее 2,4,6-тринитрофенол:
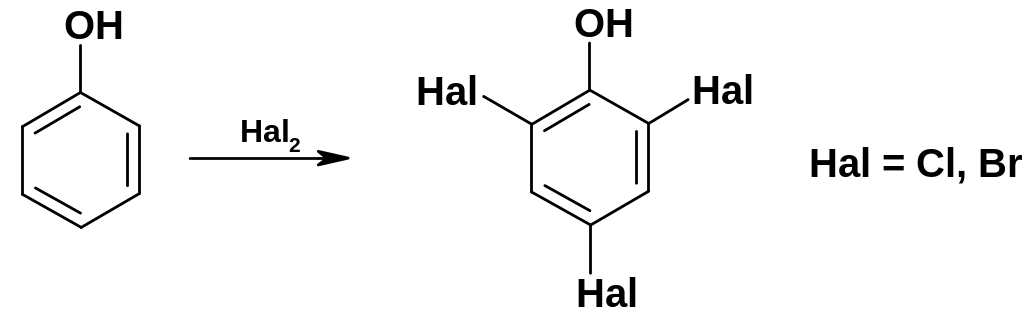 2.
Галогенирование
фенола идет исключительно легко без
катализатора;
при действии бромной или хлорной воды
замещаются три атома водорода:
2.
Галогенирование
фенола идет исключительно легко без
катализатора;
при действии бромной или хлорной воды
замещаются три атома водорода:
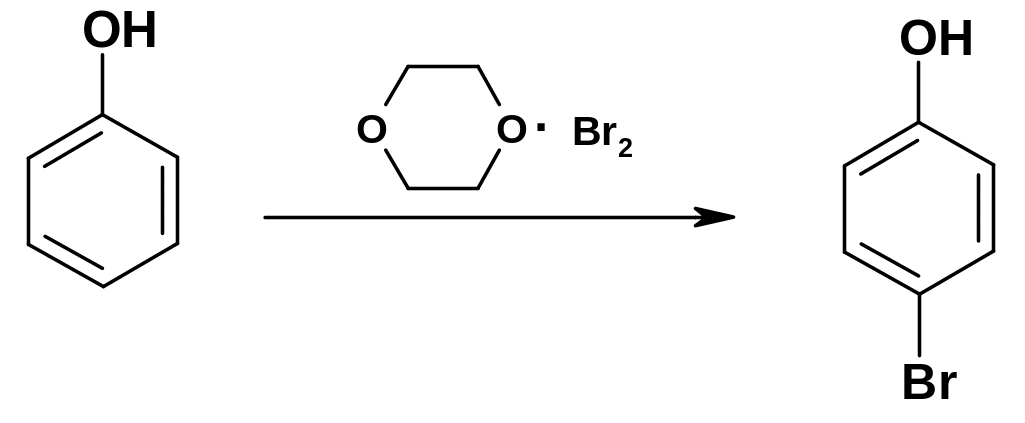 При
этом галогенируется не сам фенол, а
фенолят-анион, возникающий при его
кислотной диссоциации. Чтобы получить
моногалогенфенолы, используют более
слабые электрофильные реагенты:
сульфурилхлорид SO2С12
или комплекс диоксана с бромом –
диоксандибромид; образуются преимущественно
р-галогенфенолы:
При
этом галогенируется не сам фенол, а
фенолят-анион, возникающий при его
кислотной диссоциации. Чтобы получить
моногалогенфенолы, используют более
слабые электрофильные реагенты:
сульфурилхлорид SO2С12
или комплекс диоксана с бромом –
диоксандибромид; образуются преимущественно
р-галогенфенолы:
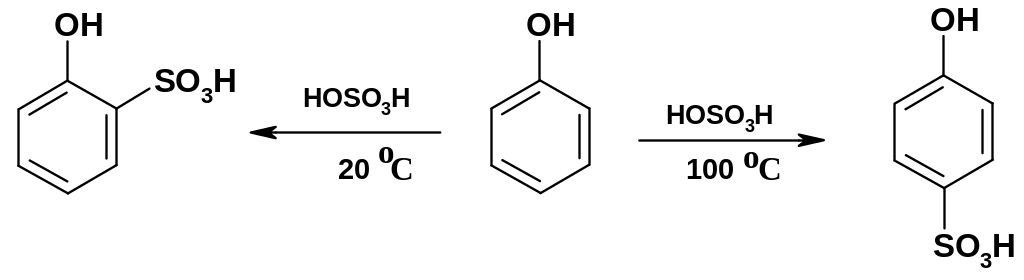 3.
Сульфирование
фенола приводит к монозамещению с
образованием фенолсульфокислот: при
20 оС
преимущественно образуется о-изомер
(кинетический контроль); при 100
оС -
преимущественно р-изомер
(термодинамический контроль; р-изомер
устойчивей о-изомера):
3.
Сульфирование
фенола приводит к монозамещению с
образованием фенолсульфокислот: при
20 оС
преимущественно образуется о-изомер
(кинетический контроль); при 100
оС -
преимущественно р-изомер
(термодинамический контроль; р-изомер
устойчивей о-изомера):
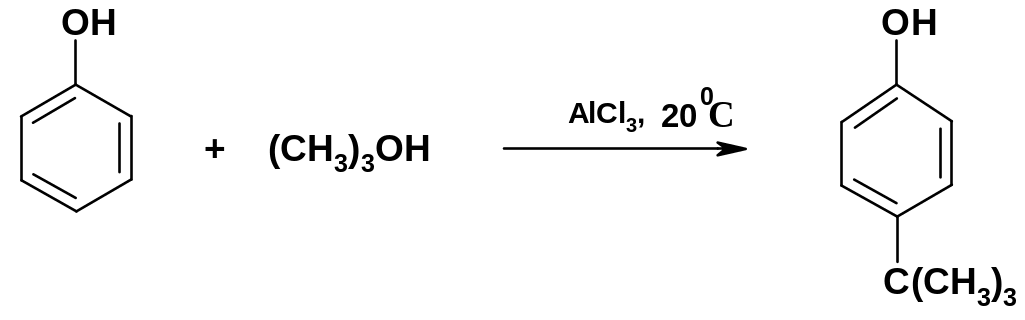 4.
Для алкилирования
фенолов в ядро (С-алкилирования) часто
используют спирты или алкены; в качестве
катализаторов используют неорганические
кислоты Льюиса, а также серную кислоту
или катионообменные смолы в Н-форме; в
большинстве случаев образуются смеси
о-
и р-алкилфенолов
и диалкилфенолов; однако в мягких
условиях можно получать индивидуальные
соединения, например, при введении в
ядро трет-бутильной
группы при 20 0С
образуется почти исключительно р-изомер:
4.
Для алкилирования
фенолов в ядро (С-алкилирования) часто
используют спирты или алкены; в качестве
катализаторов используют неорганические
кислоты Льюиса, а также серную кислоту
или катионообменные смолы в Н-форме; в
большинстве случаев образуются смеси
о-
и р-алкилфенолов
и диалкилфенолов; однако в мягких
условиях можно получать индивидуальные
соединения, например, при введении в
ядро трет-бутильной
группы при 20 0С
образуется почти исключительно р-изомер:
В качестве алкилирующих реагентов могут использоваться карбонильные соединения (альдегиды и кетоны) при кислотном катализе. Эти реакции могут идти и с обычными аренами, но особенно хорошо они протекают для фенолов. Наибольшее практическое значение имеет конденсация фенола с формальдегидом:
В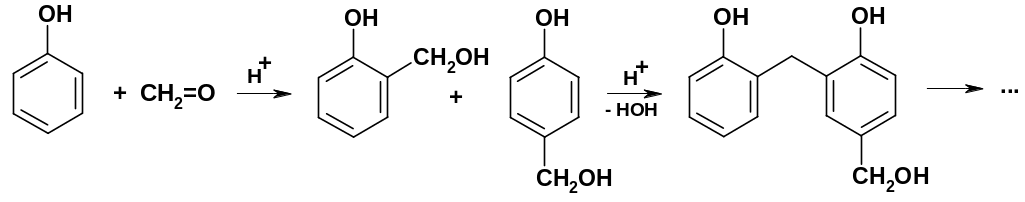 начале
образуются о-
и
р-гидроксиметильные
производные фенола; при их конденсации
между собой образуется димерный продукт;
реакция может продолжаться далее и
приводить к образованию олигомерных,
а в дальнейшем и полимерных продуктов;
фенол-формальдегидные полимеры – одни
из наиболее широко используемых
полимерных продуктов.
начале
образуются о-
и
р-гидроксиметильные
производные фенола; при их конденсации
между собой образуется димерный продукт;
реакция может продолжаться далее и
приводить к образованию олигомерных,
а в дальнейшем и полимерных продуктов;
фенол-формальдегидные полимеры – одни
из наиболее широко используемых
полимерных продуктов.
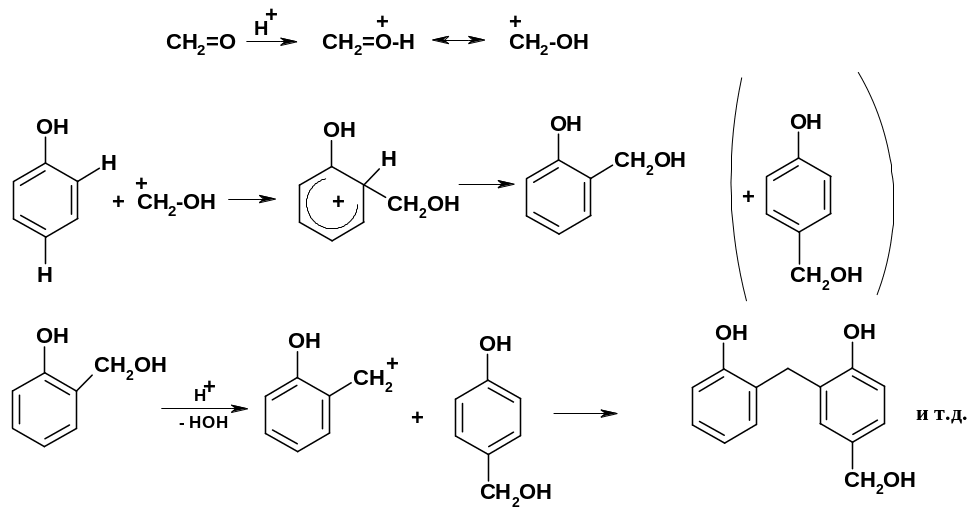
Электрофильной частицей при получении гидроксиметильных производных служит протонированная молекула формальдегида; при образовании димера происходит «спиртовое» алкилирование: одна из молекул гидроксиметильного производного выступает в роли алкилирующего спирта, а другая является субстратом:
 При конденсации
фенола с ацетоном в присутствии кислот
обычно получают продукт конденсации
двух молекул фенола с одной молекулой
ацетона – дифенилолпропан:
При конденсации
фенола с ацетоном в присутствии кислот
обычно получают продукт конденсации
двух молекул фенола с одной молекулой
ацетона – дифенилолпропан:
Э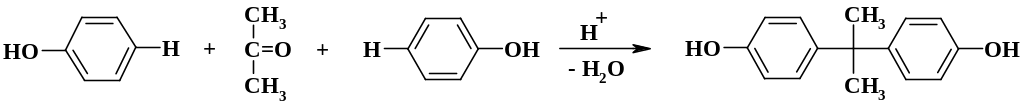 то
соединение под названием диан
используется в качестве мономера для
получения полимерных материалов.
то
соединение под названием диан
используется в качестве мономера для
получения полимерных материалов.
Своеобразным вариантом введения углеводородного радикала в ароматическое ядро фенолов является перегруппировка аллиловых эфиров фенолов, которая происходит при их нагревании; аллильная группа мигрирует в о-положение к группе ОН:
Р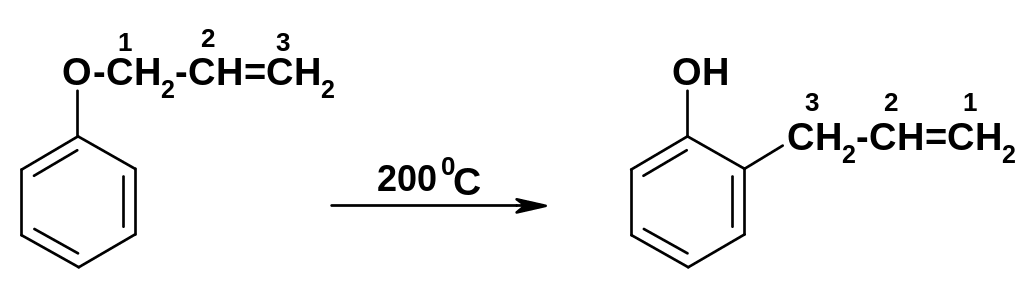 еакция
носит название перегруппировки
Клайзена.
еакция
носит название перегруппировки
Клайзена.
Использование изотопной метки показало, что в ходе миграции происходит «разворот» аллильной группы, и с ароматическим ядром связывается атом углерода С3. Это можно объяснить внутримолекулярной миграцией аллильной группы в процессе перициклической реакции – [3,3]-сигматропной перегруппировки:
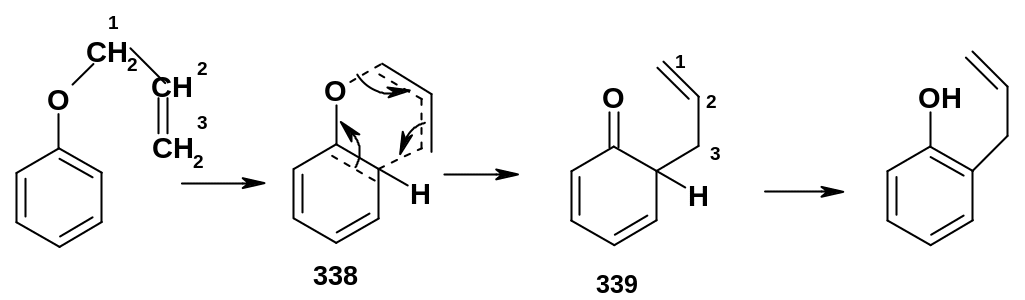 В
ходе перегруппировки образуется
шестичленное циклическое переходное
состояние (338) с делокализованной
электронной плотностью; дальнейшее
перераспределение электронов приводит
к аллильному производному циклогексадиенона
– кето-форме о-аллил-
фенола (339), которая немедленно
перегруппировывается в более стабильный
о-аллилфенол.
В
ходе перегруппировки образуется
шестичленное циклическое переходное
состояние (338) с делокализованной
электронной плотностью; дальнейшее
перераспределение электронов приводит
к аллильному производному циклогексадиенона
– кето-форме о-аллил-
фенола (339), которая немедленно
перегруппировывается в более стабильный
о-аллилфенол.
5. Ацилирование фенолов в ядро можно проводить двумя путями:
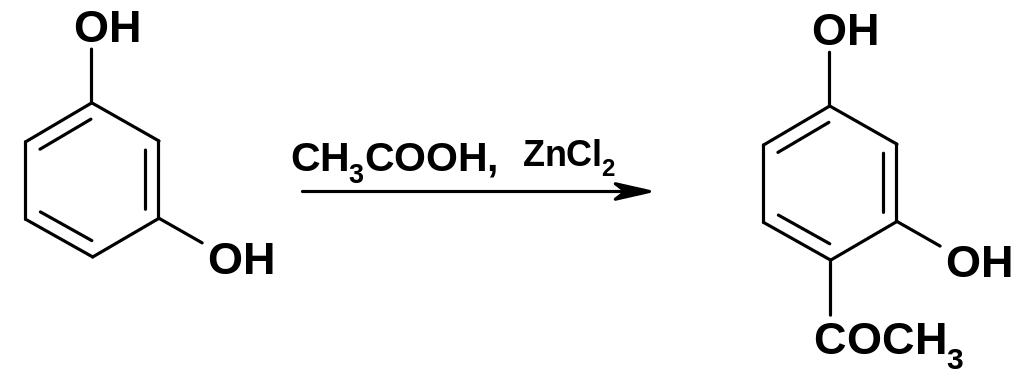 а.
Прямое
ацилирование
галогенангидридами карбоновых кислот
в присутствии кислот Льюиса (А1С13,
TiCl4);
при этом ацильная группа вступает в о-
и р-положения
к группе ОН (стр. 134). Для ацилирования
более активных субстратов, в частности,
двухатомных фенолов, можно использовать
и более слабые ацилирующие реагенты,
например, карбоновые кислоты:
а.
Прямое
ацилирование
галогенангидридами карбоновых кислот
в присутствии кислот Льюиса (А1С13,
TiCl4);
при этом ацильная группа вступает в о-
и р-положения
к группе ОН (стр. 134). Для ацилирования
более активных субстратов, в частности,
двухатомных фенолов, можно использовать
и более слабые ацилирующие реагенты,
например, карбоновые кислоты:
Прямое ацилирование фенолов часто протекает с осложнениями; в этих случаях лучшие результаты могут быть получены при непрямом ацилировании – перегруппировке сложных эфиров фенолов.
б. Перегруппировка сложных эфиров фенолов (перегруппировка Фриса).
П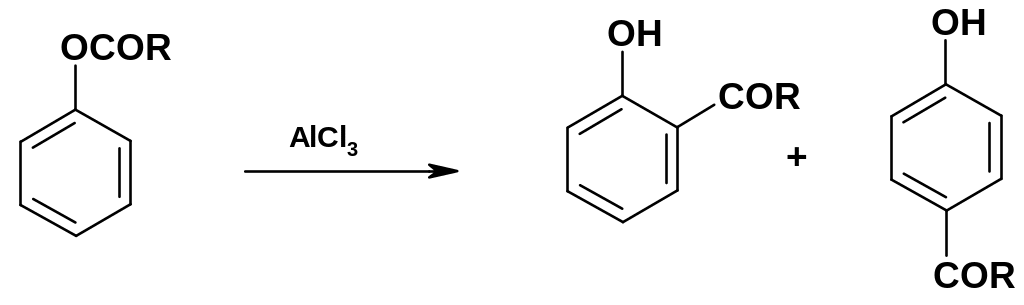 ри
непрямом ацилировании вначале получают
сложный эфир фенола (проводят
О-ацилирование); при действии на сложный
эфир фенола хлорида алюминия происходит
перегруппировка с миграцией ацильной
группы в о-
и р-положения
к группе ОН:
ри
непрямом ацилировании вначале получают
сложный эфир фенола (проводят
О-ацилирование); при действии на сложный
эфир фенола хлорида алюминия происходит
перегруппировка с миграцией ацильной
группы в о-
и р-положения
к группе ОН:
Эту реакцию называют перегруппировкой Фриса. Миграция ацильной группы идет межмолекулярно – в отличие от рассмотренной выше перегруппировки Клайзена.
Реакцию можно направлять в сторону получения одного изомера; в частности, при низких температурах преобладают р-изомеры, а при повышенных (~100 оС) – о-изомеры.
Частный случай ацилирования - реакции формилирования – введение формильной (альдегидной) группы в ароматическое ядро. Ранее было рассмотрено формилирование аренов по Гаттерману-Коху (стр. 131). Для фенолов становится возможным формилирование при помощи более мягких реагентов.
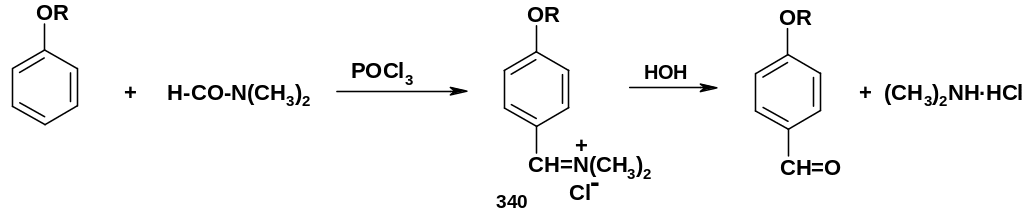 а.
Формилирование
по Вильсмайеру
(иногда называется формилированием по
Вильсмайеру-Хааку): действие на фенолы
или их простые эфиры смеси диметилформамида
(ДМФА) и хлороксида фосфора:
а.
Формилирование
по Вильсмайеру
(иногда называется формилированием по
Вильсмайеру-Хааку): действие на фенолы
или их простые эфиры смеси диметилформамида
(ДМФА) и хлороксида фосфора:
Первичным продуктом реакции является иминиевая соль (340), которая при гидролизе превращается в альдегид.
Реакция образования продукта (340) протекает следующим образом:
Х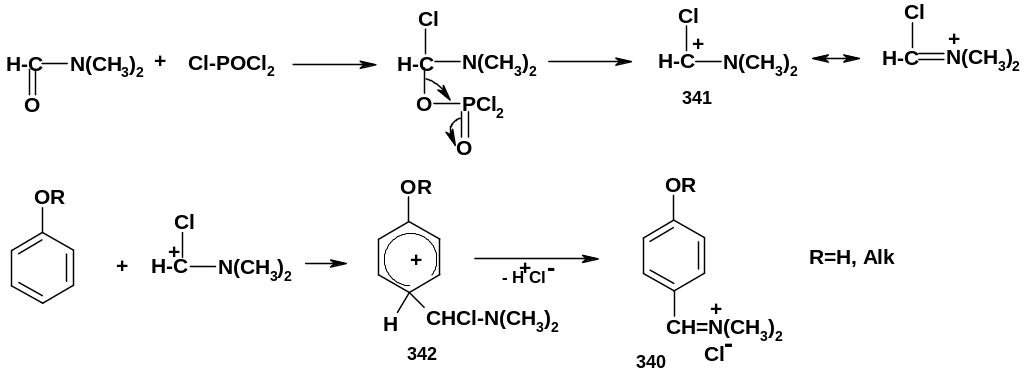 лороксид
фосфора присоединяется по связи С=О
диметилформамида; далее от аддукта
отщепляется фосфатный фрагмент и
образуется хлориминиевый катион (341);
он и является электрофильной частицей,
атакующей ароматическое ядро фенола
(эфира) с образованием -аддукта
(342), из которого и образуется соединение
(340). Катион (341) стабилизирован резонансом,
поэтому он устойчив и сравнительно
малоактивен; обычные одноядерные арены
по Вильсмайеру не формилируются (хотя
более активный антрацен формилируется
в положение 9). Альдегидная группа
вступает, в основном, в пара-положение
к группе OН
(OR),
хотя возможно и образование орто-изомера.
лороксид
фосфора присоединяется по связи С=О
диметилформамида; далее от аддукта
отщепляется фосфатный фрагмент и
образуется хлориминиевый катион (341);
он и является электрофильной частицей,
атакующей ароматическое ядро фенола
(эфира) с образованием -аддукта
(342), из которого и образуется соединение
(340). Катион (341) стабилизирован резонансом,
поэтому он устойчив и сравнительно
малоактивен; обычные одноядерные арены
по Вильсмайеру не формилируются (хотя
более активный антрацен формилируется
в положение 9). Альдегидная группа
вступает, в основном, в пара-положение
к группе OН
(OR),
хотя возможно и образование орто-изомера.
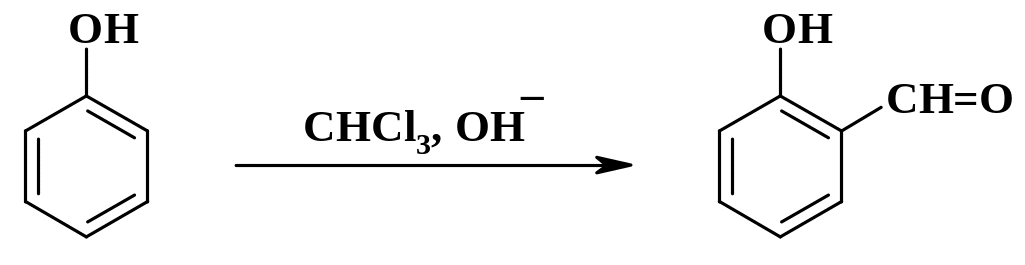 б.
Формилирование
по Раймеру-Тиману.
Реакция происходит при действии на
фенолы хлороформа и щелочи; формилирование
происходит по орто-положению:
б.
Формилирование
по Раймеру-Тиману.
Реакция происходит при действии на
фенолы хлороформа и щелочи; формилирование
происходит по орто-положению:
Э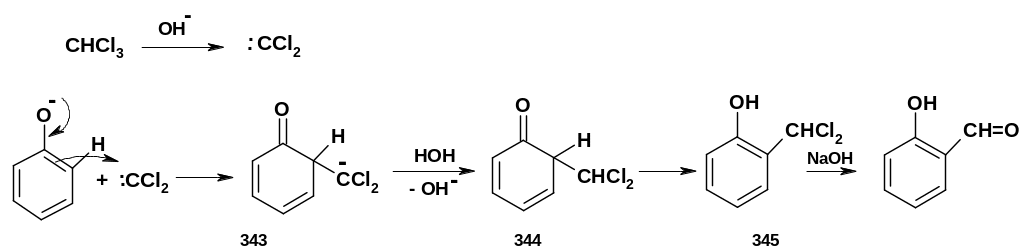 лектрофильной
частицей здесь является дихлоркарбен,
который образуется при действии щелочи
на хлороформ (-элиминирование,
стр. 167):
лектрофильной
частицей здесь является дихлоркарбен,
который образуется при действии щелочи
на хлороформ (-элиминирование,
стр. 167):
Дихлоркарбен взаимодействует с фенолят-анионом (который образуется из фенола в щелочной среде); образующийся анион (343) отрывает протон от воды с образованием соединения (344) – кето-формы о-замещенного фенола, которое немедленно перегруппировывается в фенол (345); гидролиз дихлорпроизводного приводит к о-гидроксиальдегиду.
Реакции Вильсмайера и Раймера-Тимана хорошо дополняют друг друга, поскольку первая ориентирует формильную группу преимущественно в р-положение, а вторая – преимущественно в о-положение.
Б. Новые электрофильные реакции, не протекающие с аренами
1. Карбоксилирование фенолятов (реакция Кольбе-Шмитта). При нагревании фенолятов в атмосфере СО2 под давлением образуются соли фенолкарбоновых кислот: при Т < 200 оС – преимущественно орто-изомеры, при Т > 200 оС – преимущественно пара-изомеры. Таким образом, происходит прямое введение карбоксильной группы в ароматическое ядро. Наиболее известный пример – получение о-гидроксибензойной (салициловой) кислоты:
О бразующуюся
соль подкисляют и получают салициловую
кислоту.
бразующуюся
соль подкисляют и получают салициловую
кислоту.
 Фенолят
–ион настолько активен в реакциях
электрофильного замещения, что реагирует
даже с таким весьма слабым электрофилом,
как СО2.
Детали механизма не до конца ясны, но
это несомненно электрофильное замещение.
Это подтверждается, в частности, тем,
что если в ядре присутствуют дополнительные
электронодонорные группы, реакция
протекает еще легче: например,
мета-аминофенолят
натрия карбоксилируется уже при кипячении
с раствором NaHCO3;
образуется пара-аминосалициловая
кислота:
Фенолят
–ион настолько активен в реакциях
электрофильного замещения, что реагирует
даже с таким весьма слабым электрофилом,
как СО2.
Детали механизма не до конца ясны, но
это несомненно электрофильное замещение.
Это подтверждается, в частности, тем,
что если в ядре присутствуют дополнительные
электронодонорные группы, реакция
протекает еще легче: например,
мета-аминофенолят
натрия карбоксилируется уже при кипячении
с раствором NaHCO3;
образуется пара-аминосалициловая
кислота:
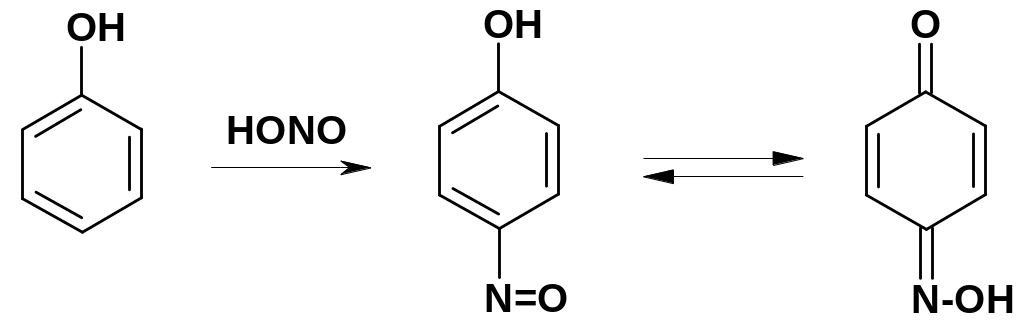 2.
Реакции
нитрозирования.
При действии на фенолы азотистой кислоты
(смеси NaNO2
и НCl)
происходит замещение атома водорода в
пара-положении
к группе ОН на нитрозогруппу NO:
2.
Реакции
нитрозирования.
При действии на фенолы азотистой кислоты
(смеси NaNO2
и НCl)
происходит замещение атома водорода в
пара-положении
к группе ОН на нитрозогруппу NO:
Вероятно, электрофильной частицей в этой реакции является катион +N=О - довольно малоактивный электрофил.
Продукт реакции – р-нитрозофенол – находится в таутомерном равновесии с монооксимом р-хинона (хиноны будут рассмотрены далее).
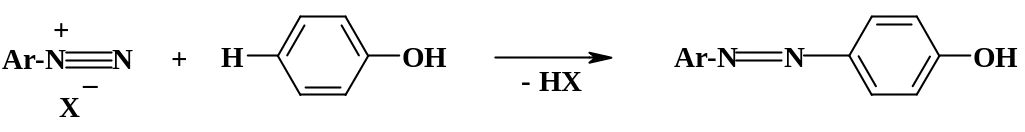 3.
Реакции
азосочетания
происходят при взаимодействии фенолов
с ароматическими диазосоединениями;
образуются ароматические
азосоединения
(азоарены)
3.
Реакции
азосочетания
происходят при взаимодействии фенолов
с ароматическими диазосоединениями;
образуются ароматические
азосоединения
(азоарены)
Более подробно эта реакция будет рассматриваться в разделе «Диазосоединения».
Реакции со слабыми электрофилами, кроме фенолов, характерны для ароматических аминов, т.к. аминогруппа, подобно гидроксигруппе обладает сильным +М-эффектом и активирует ядро к электрофильным реакциям (см. далее в главе «Амины»).
IV.3.2.3. Реакции окисления и восстановления фенолов.
А. Реакции окисления. Ароматическое ядро фенолов, имеющее повышенную электронную плотность, окисляется значительно легче, чем ароматическое ядро аренов. При стоянии на воздухе фенолы темнеют, жидкие – быстрее, кристаллические – медленнее. Окисление фенолов протекает по разным направлениям, иногда весьма сложно.
1. Окисление одноатомных фенолов. Наиболее характерно окисление одноатомных фенолов одноэлектронными окислителями, например, K3Fe(CN)6 в щелочной среде. Эти окислители, отнимая один электрон, превращают фенолят-анионы (возникающие в щелочной среде) в радикалы.
В большинстве случаев эти радикалы далее рекомбинируют по нескольким схемам (приведен один вариант рекомбинации):
Т аким
образом, результатом реакции является
окислительное
сочетание
(окислительная димеризация) фенолов.
аким
образом, результатом реакции является
окислительное
сочетание
(окислительная димеризация) фенолов.
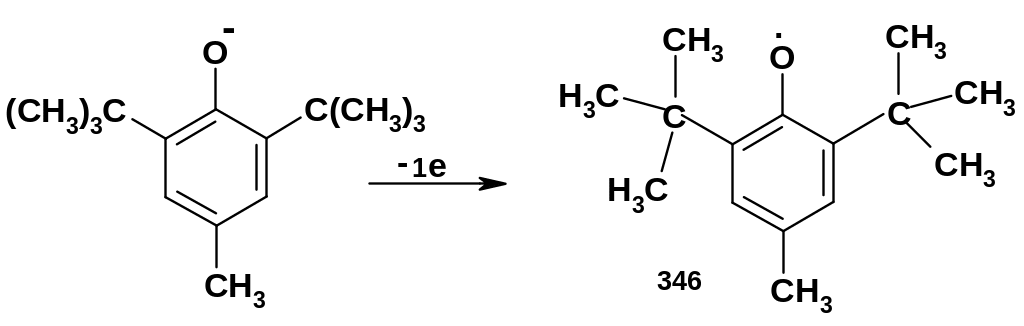 Однако
в некоторых случаях рекомбинация
образующихся радикалов затруднена; это
особенно свойственно таким радикалам,
у которых радикальный центр на кислороде
пространственно экранирован объёмными
орто-заместителями,
и, к тому же, занято пара-положение;
примером является радикал (346), образующийся
из 2,6-дитрет-бутил-4-метилфенола
(точнее из его фенолята):
Однако
в некоторых случаях рекомбинация
образующихся радикалов затруднена; это
особенно свойственно таким радикалам,
у которых радикальный центр на кислороде
пространственно экранирован объёмными
орто-заместителями,
и, к тому же, занято пара-положение;
примером является радикал (346), образующийся
из 2,6-дитрет-бутил-4-метилфенола
(точнее из его фенолята):
Рекомбинация радикала (346) ( и его аналогов) крайне затруднена по стерическим причинам (отталкивание между объёмными группами при рекомбинации); поэтому радикалы такого типа являются долгоживущими. Данный тип долгоживущих радикалов называют ароксильными долгоживущими радикалами. Существует несколько других типов долгоживущих радикалов: триарилметильные (стр. 144), нитроксильные и др.; долгоживущие радикалы имеют не только теоретическое, но и заметное практическое значение; в частности, они могут ингибировать нежелательные радикальные процессы, например, окисление. В частности, 2,6-дитрет-бутил-4-метилфенол, из которого образуется радикал (346), под названием ионол использовался как антиоксидант в пищевых продуктах.
2. Окисление двухатомных и многоатомных фенолов. Увеличение числа гидроксильных групп в ароматическом ядре еще более ускоряет окисление фенолов. Например, 1,2,3-тригидроксибензол (пирогаллол) окисляется кислородом воздуха настолько быстро, что его щелочные растворы могут использоваться для удаления кислорода из воздуха.
 Наиболее
практически важным является окисление
двухатомных фенолов с гидроксильными
группами в орто-
или в пара-
положениях; продуктами окисления
являются соответственно орто-
или пара-хиноны
(347) и (348):
Наиболее
практически важным является окисление
двухатомных фенолов с гидроксильными
группами в орто-
или в пара-
положениях; продуктами окисления
являются соответственно орто-
или пара-хиноны
(347) и (348):
В качестве окислителей используют бихроматы, оксид серебра, иминоксилдисульфонат калия (КО3S)2N-O· («соль Фреми») и др. Простые эфиры двухатомных фенолов также можно окислить до хинонов; здесь используют церийаммонийнитрат Се(NH4)2(NO2)6.
Хиноны – ярко окрашенные соединения со специфическими свойствами, они используются для производства красителей; многие хиноны обладают высокой биологической активностью. Хиноны будут рассмотрены в разделе «Карбонильные соединения».
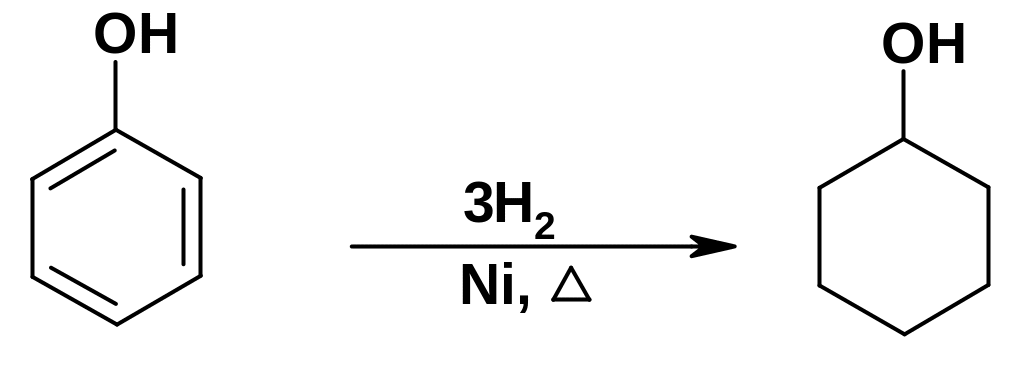 Б.
Восстановление
фенолов.
Ароматическое ядро одноатомных
фенолов восстанавливается несколько
труднее, чем в случае аренов; тем не
менее, в присутствии металлов платиновой
группы при повышенной температуре
гидрирование ядра происходит. Наибольшее
практическое значение имеет гидрирование
самого фенола до циклогексанола:
Б.
Восстановление
фенолов.
Ароматическое ядро одноатомных
фенолов восстанавливается несколько
труднее, чем в случае аренов; тем не
менее, в присутствии металлов платиновой
группы при повышенной температуре
гидрирование ядра происходит. Наибольшее
практическое значение имеет гидрирование
самого фенола до циклогексанола:
Циклогексанол далее используется для синтеза важного мономера – капролактама.
Значительно легче восстанавливаются двухатомные фенолы с группами ОН в мета-положении: восстановление идет уже при действии амальгамы натрия; продуктами восстановления являются дигидропроизводные (349), которые находятся в таутомерном равновесии с моноенольными производными (350) и циклическими 1,3-дикетонами (351):
В равновесной смеси присутствуют в
основном формы (350) и (351). Циклические
1,3-дикетоны (как и нециклические) проявляют
свойства достаточно сильных СН-кислот
и благодаря этому интенсивно используются
в органическом синтезе в реакциях,
сопровождающихся увеличением углеродного
скелета. 1,3-Дикетоны будут рассмотрены
позднее.
равновесной смеси присутствуют в
основном формы (350) и (351). Циклические
1,3-дикетоны (как и нециклические) проявляют
свойства достаточно сильных СН-кислот
и благодаря этому интенсивно используются
в органическом синтезе в реакциях,
сопровождающихся увеличением углеродного
скелета. 1,3-Дикетоны будут рассмотрены
позднее.