

2 Електрохімічна корозія
2.l Виникнення електродного потенціалу
Електрохімічна корозія металів представляє собою самовільне руйнування металу внаслідок електрохімічного взаємодії його з електролітом. Це гетерогенна реакція, що протікає на поверхні металу. Причиною електрохімічної корозії є термодинамічна нестійкість металу в даних корозійних умовах.
Для розуміння механізму електрохімічної корозії необхідно встановити, які процеси спостерігаються на межі метал - розчин. На межі розділення двох фаз при певних умовах може виникнути різниця потенціалів або, як прийнято говорити, стрибок потенціалу(наприклад електродний потенціал - на межі метал - розчин; контактний потенціал - на межі двох різних металів; контактний потенціал другого роду - на межі метал - газ; дифузійний потенціал - на кордоні двох розчинів), що мають різну концентрацію розчиненої речовини, та ін.
Стрибок потенціалу між двома фазами визначається переходом заряджених часток з однієї фази в іншу або вибірковою адсорбцією заряджених або полярних частинок однієї фази на поверхні іншої з утворенням подвійного електричного шару.
Розглянемо механізм виникнення електродного потенціалу. Електродом називається метал, занурений у розчин електроліту. Що ж відбувається на поверхні металу при зануренні його в розчин власних іонів?
У металі і в розчині є однакові іони - іони металу. У металі ці іони перебувають у вузлах кристалічної решітки і утримуються в ній завдяки енергії зв'язку іонів решітки. Щоб вивести іон з кристалічної решітки, необхідно затратити роботу, рівну енергії зв'язку іонів, яку також можна назвати роботою виходу іона з металу.
В розчині іони металу оточені полярними молекулами води, тобто перебувають у гідратованому стані. Щоб вивести іон металу з розчину необхідно виконати роботу, рівну енергії гідратації, тобто енергії зв'язку іона металу з молекулами води.
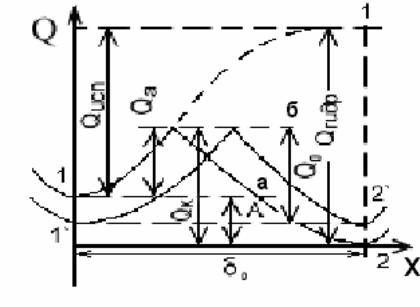
Рисунок 2.1. Схема зміни енергії при випаровуванні катіона металу у вакуум і при переході в розчин:
а - момент занурення металу в розчин його солі; б - момент встановлення рівноваги.
Встановлення електродного потенціалу на металі залежить від співвідношення енергії кристалічної решітки та енергії гідратації іонів. Знаходяться на поверхні металу катіони мають запас потенційної енергії, що відповідає значенню енергії в точці 1 (нижня) на рис. 2.1. Відрив катіона від поверхні металу з переходом у вакуум вимагає значної енергії (пунктирна крива 1-1), відповідної енергії випаровування Qвип. Полярні молекули води (або іншого розчинника), орієнтуючись навколо поверхневих катіонів металу, полегшують перехід катіонів у розчин із звільненням енергії гідратації, тому що рівень енергії гідратованого іону нижче, ніж катіона у вакуумі, на величину Qгідр. Потенційна енергія катіонів, що знаходяться в розчині, в межах подвійного електричного шару відповідає точці 2. Для переходу в розчин поверхневий катіон металу повинен подолати лише енергетичний бар'єр Qа. Різниця рівнів потенційних енергій в точках 1 і 2, що дорівнює А, відповідає роботі процесу переходу іонів металу в розчин. Для переходу з розчину в метал гідратований катіон повинен подолати енергетичний бар'єр Qк. Відповідно до теорії О.М.Фрумкіна, при взаємодії металу і розчину протікають два сполучених процеси:
1. Перехід іонів з металу в розчин з утворенням гідратованих іонів (анодний процес):
Me + mН2O = Меn+ · mН2O + ne.
Швидкість цього процесу, виміряна числом іонів, що переходять з однієї фази в іншу через одиницю поверхні за одиницю часу, може бути виражена через щільність струму ir.
2. Перехід іонів з розчину з виділенням їх на поверхні металу у вигляді нейтральних атомів, що входять до складу кристалічної решітки металу (катодний процес):
Меn+ · mН2O + ne = Ме + mН2O.
Швидкість катодного процесу виражається через відповідну щільність струму is. Який з цих процесів переважає, визначається рівнем потенційної енергії катіонів у вузлах кристалічної решітки металу UМе і в розчині Uр.
Якщо UМе > Uр , то переважає анодний процес, сумарна швидкість якого iа =ir – is. Розчин отримує надлишковий позитивний заряд у вигляді катіонів металу, а поверхня металу набуває надлишковий негативний заряд за рахунок надлишку електронів. Перехід частини катіонів у розчин супроводжується зниженням середньої потенційної енергії поверхневих катіонів (точка 1 переміщається вниз до точки 1'), появою на металевій поверхні надлишкових негативних зарядів і підвищенням енергетичного бар'єру Qа. Підвищення концентрації іонів біля поверхні металу супроводжується зростанням потенційної енергії (точка 2 переміщається вгору до точки 2'), придбанням розчином надлишкового позитивного заряду і зниженням енергетичного бар'єру Qк. По мірі збільшення концентрації катіонів біля поверхні, із зростанням величини заряду розчину і металу, протікання прямого процесу ускладнюється і полегшується перебіг зворотного процесу, тобто перехід іонів металу з розчину в кристалічну решітку.
Коли енергетичні рівні іонів на поверхні металу і в розчині стають однаковими, тобто UМе = Uр, встановлюється динамічна рівновага. Рівновага характеризується тим, що Qк = Qа = Qо і швидкості анодного і катодного процесів є рівними: ir = is = iо, де iо - щільність струму обміну.
Внаслідок електростатичного притягання катіонів та надлишкових електронів на поверхні металу іони металу не можуть віддалитися вглиб розчину, а перебувають біля поверхні. Утворюється подвійний електричний шар (рис.2.2, а).Різниця електричних потенціалів, що виникає на межі метал - розчин, внаслідок надлишкових зарядів, називається електродним потенціалом.
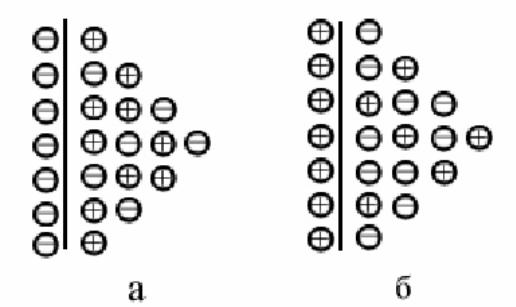
Рисунок 2.2. Подвійний електричний шар системи: а – Zn/ZnSO4; б – Cu/CuSO4.
Якщо UМе ˂ Uр, то потенційна енергія катіона в металі менше потенційної енергії гідратованого іону. Тоді в початковий момент відбувається не розчинення, а осадження металу з розчину, сумарна швидкість якого ік = is – ir. При цьому поверхня металу набуває надлишковий позитивний заряд катіонів, a надлишок аніонів що залишився в приелектродному просторі надає йому негативний заряд. Позитивний заряд поверхні металу ускладнює подальше осадження катіонів і полегшує зворотний процес - перехід іонів металу з решітки в розчин. В результаті в системі встановлюється динамічна рівновага і виникає подвійний електричний шар з протилежним розподілом зарядів, тобто поверхня металу заряджається позитивно, а в розчині у поверхні металу утворюється надлишок аніонів (рис.2.2, б).Таким чином, незалежно від енергетичних співвідношень, настає динамічна рівновага між металом і розчином його солі, яка характеризується певною за величиною і знаком рівноважним потенціалом.
2.2 Рівноважні і не рівноважні електродні потенціали
При зануренні металів в розчин будь-якого електроліту виникає електродний потенціал. Якщо у встановленні потенціалу приймають участь тільки власні іони металу (заряди переносяться тільки ними), то такий потенціал називають рівноважним, або оборотним (рис. 2.3, а). Величина рівноважного електродного потенціалу залежить від природи металу і розчинника, температури, активності іонів металу в розчині. Рівноважні потенціали металів, визначені для активності іонів металу в розчині, що дорівнює одиниці, при температурі 25 °С, називають стандартними електродними потенціалами. Стандартні електродні потенціали можна розрахувати по зміні ізобарно - ізотермічних потенціалів електродних процесів, віднесених до 1 моль металу і виражених у вольтах:
ΔG = – nFE; E = – (ΔG/nF). (2.1)
За відомим значенням енергії Гіббса реакції (ΔG) можна розрахувати величину електродного потенціалу. Рівняння (2.1) показує перетворення хімічної енергії в електричну та назад.
Значення стандартних електродних потенціалів для деяких металів приведені в табл.2.1.
Ряд металів, розташований за зростанням позитивних значень стандартних електродних потенціалів, називається рядом напруг.
Положення металу в ряду визначає його хімічну активність, окисні і відновні властивості. Чим більш негативне значення потенціалу має метал, тим більшою мірою зростає його здатність до окислення.
Таблиця 2.1
Електрод |
K+ | K↔ |
Al3+|Al↔ |
Ti2+|Ti↔ |
Mn2+|Mn↔ |
Zn2+|Zn↔ |
↔Cr3+|Cr |
E0, В |
- 2,925 |
- 1,66 |
- 1,63 |
- 1,18 |
- 0,762 |
- 0,74 |
Електрод |
Fe2+|Fe |
2H+|H2↔ |
Cu2+|Cu |
Hg2+|Hg |
Pd2+|Pd |
Pt2+|Pt↔ |
E0, В |
- 0,447 |
0,000 |
+ 0,337 |
+ 0,799 |
+ 0,987 |
+ 1,19 |
Залежність рівноважного електродного потенціалу від активності іонів металу в розчині і температури визначається формулою Нернста:
Е + Е0 + (RT / nF)∙lnaМеn+ , (2.2)
де E0 - стандартний електродний потенціал металу, В, Т - температура вимірювання потенціалу, К; n - ступінь окислення металу; F - число Фарадея, 96500 Кл; R - універсальна газова стала, 8,31 Дж / К; а - активність іонів металу в розчині, г-іон / л.
Підставивши значення всіх констант (при Т = 298 К) в формулу 2.2, одержимо:
E + E0 (0,059 / n)lgaМеn+ (2.3)
Приклад. Визначити потенціал мідного електроду в розчині CuSO4, якщо Cu2+ = 0,001 г-іон / л, E0 = + 0,337 В: E = 0,337 + (0,059 / 2) lg 10-3 = 0,337 - 0,088 = = + 0,249 (В).
У багатьох практичних випадках на металах (мідь, ртуть, срібло) встановлюються рівноважні, або оборотні, потенціали. Абсолютні значення стандартних потенціалів визначити експериментально і обчислити теоретично не представляється можливим. У зв'язку з цим їх визначають по відношенню до стандартного водневого електрода, потенціал якого умовно прийнятий рівним нулю.
Якщо у встановленні електродного потенціалу приймають участь не тільки власні іони металу, але й інші іони і атоми, то виникають не рівноважні, чи необоротні, потенціали. Умовою утворення не рівноважного потенціалу є рівність швидкостей переносу зарядів у прямому і зворотному напрямках, тобто баланс заряду, але баланс маси при цьому не дотримується, так як у передачі зарядів беруть участь різні часточки (рис. 2.3, б).
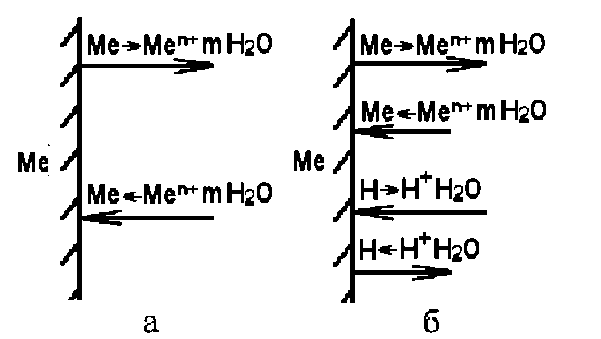
Рисунок 2.3. Схема встановлення потенціалів: а - рівноважного; б - не рівноважного.
Стале в часі значення незворотного електродного потенціалу металу, відповідне рівності сум швидкостей анодних і катодних процесів, називають стаціонарним потенціалом металу. Метал переважно розчиняється, а баланс зарядів, які переносяться у зворотному напрямку, компенсується іонами металу та іншими частками, наприклад переходом Н+ з розчину в газову фазу.
Таким чином, при встановленні на металі незворотного електродного потенціалу може відбуватися електрохімічне розчинення металу:
Me + mH2O → Men+1∙mH2O + ne (анодний процес)
і відновлення будь-якого деполяризатора (іона або молекули), що знаходиться в розчині, наприклад, іонів водню:
H+H2O + e → H + H2O (катодний процес).
До необоротних електродних потенціалів відносяться потенціали багатьох металів у розчинах власних іонів (нікель, залізо, хром, титан та ін.) Для не рівноважних потенціалів формула Нернста непридатна, тому що електродний потенціал визначається декількома паралельними реакціями.
Величини необоротних електродних потенціалів металів залежать як від внутрішніх чинників, пов'язаних з природою металу, так і від зовнішніх, пов'язаних зі складом електроліту і фізичними умовами. До внутрішніх факторів належать: фізико-хімічний стан і структура металу, стан поверхні, наявність механічних деформацій і напружень та ін. Зовнішні чинники - це хімічна природа розчинника, природа і концентрація розчинених газів, температура, тиск, перемішування розчину та інші. Не рівноважні електродні потенціали визначаються тільки дослідним шляхом. У табл. 2.2 наведено дослідні значення стаціонарних потенціалів ряду металів в нейтральних середовищах.
Таблиця 2.2
Потенціал |
Al |
Zn |
Cr |
Fe |
Ni |
Cu |
E0, В |
- 1,66 |
- 0,762 |
- 0,74 |
- 0,44 |
- 0,23 |
+ 0,337 |
Eстац. 3% NaCl |
- 0,63 |
- 0,83 |
+ 0,23 |
- 0,50 |
- 0,02 |
+ 0,05 |
Eстац. 1% Na2SO4 |
- 0,47 |
- 0,81 |
- |
- 0,5 |
+ 0,035 |
+ 0,24 |
Для вирішення питання, чи є потенціал даного металу в будь-якому електроліті оборотним або незворотнім, слід зіставити теоретичне значення, розраховане за рівнянням (2.1), і значення електродного потенціалу металу, отримане дослідним шляхом.
2.3 Будова подвійного електричного шару
Як було показано раніше, на межі метал - розчин утворюється подвійний електричний шар. Іони, що зібралися біля поверхні металу при встановленні рівноваги, не можуть покинути приелектродний шар і віддалитися від нього углиб розчину. Цьому перешкоджає електростатичне притягання між іонами і надлишковим зарядом на поверхні металу. Однак подвійний електричний шар може утворитися і без переходу заряджених частинок з фази у фазу. У цьому випадку утворення подвійного шару можливо за рахунок вибіркової адсорбції іонів однієї фази на поверхні іншої, наприклад специфічна адсорбція аніонів хлору з водного розчину солі на поверхні будь-якого інертного металу. Це призводить до появи в прикатодному шарі надлишкового негативного заряду і позитивного заряду в прилеглому шарі розчину.
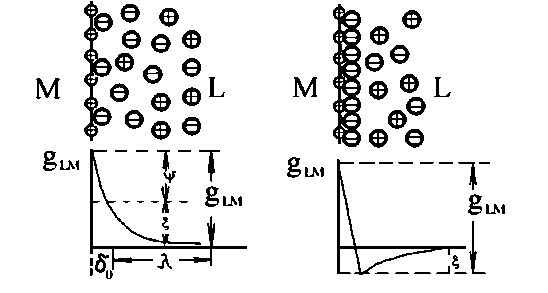
а б
Рисунок 2.4. Будова подвійного електричного шару: а - при відсутності в розчині поверхнево-активних речовин; б - при їх наявності.
Будова подвійного шару і зміна потенціалу із збільшенням відстані від поверхні металу для розчинів, що не містять поверхнево-активних речовин, показані на рис. 2.4 а; для розчинів, що містять поверхнево-активні аніони - на рис. 2.4, б. Відповідно до теорії Штерна, подвійний електричний шар поділяється нa щільну частину δ0, товщина якої дорівнює середньому радіусу іонів електроліту, і дифузійну частину, де концентрація іонів поступово падає, досягаючи концентрації, властивої цьому розчину в цілому.
Величина електродного потенціалу складається з потенціалів щільної ψψ і дифузної ψζ' частин подвійного шару: gLM = ψ ψψ + ζ'ψ'. Загальна товщина подвійного шару складається з товщини, приблизно рівною радіусу сольватованого іону (щільна частина подвійного шару), і товщини дифузійної частини подвійного шару: μ = δ0 + λ.
Товщина дифузійної частини подвійного шару залежить від природи і особливо від концентрації розчину. У чистій воді товщина дифузної частини зменшується, а з підвищенням температури, внаслідок теплового руху розчину - збільшується.
За теорією Штерна, в щільній частині подвійного шару іони утримуються не тільки електростатичними силами, але і силами специфічної адсорбції, тобто силами некулонівского походження. Тому в розчинах, що містять поверхнево - активні іони, їх число в щільній частини подвійного шару може бути не еквівалентним заряду поверхні металу, а перевершувати його на деяку величину, що залежить від властивостей іонів і заряду металу.
Таким чином, слід розрізняти дві моделі подвійного електричного шару, одна з яких належить до розчинів, що не містять поверхнево активних речовин (рис.2.4, а), інша - до розчинів, що містить специфічно адсорбовані іони (рис.2.4, б).
2.4. Потенціал нульового заряду
Електрод з аніонним подвійним електричним шаром має позитивний потенціал щодо розчину (рис.2.5, а).

а б в
Рисунок 2.5. Схема перезарядки поверхні металу при катодній поляризації:
а - позитивний заряд; б - відсутність заряду; в - негативний заряд.
Якщо такий електрод піддати катодній поляризації, тобто послати на нього від зовнішнього джерела струму негативні заряди (електрони), то можна досягти зникнення на його поверхні надлишку позитивних зарядів, при цьому зникне і аніонний подвійний електричний шар (рис. 2.5, б). Потенціал, при якому поверхня металу не заряджена (відсутній іонний подвійний електричний шар), називають, по А. Н. Фрумкіна, потенціалом нульового заряду Eq = 0. При подальшій катодній поляризації металу відбувається перезарядка його поверхні з утворенням відповідного катодного електричного шару (рис. 2.5, в).
Л. І.Антропов запропонував замість загального поняття «потенціал нульового заряду» два нові терміна: потенціал незарядженої поверхні Eq = 0 і нульова точка EN. Потенціал незарядженої поверхні для даного металу і розчинника змінюється в широких межах залежно від природи і концентрації речовин, присутніх у розчині. Нульова точка - значення потенціалу незарядженої поверхні в розчині, не містить ніяких поверхнево - активних частинок. Це значення є константою, характерною для даного металу і даного розчинника. Нульова точка і потенціал незарядженої поверхні знаходяться між собою приблизно в такому ж співвідношенні, як стандартний і рівноважний потенціали електроду.
Джерелом ЕРС між металами при потенціалах нульових зарядів, з теорії А. М. Фрумкіна, може бути контактна різниця потенціалів, а також адсорбція іонів і полярних молекул. Різниця потенціалів нульових зарядів двох металів повинна бути приблизно рівною контактному потенціалу між ними (так званого контактного потенціалу Вольта):
ENMe1 − ENMe2 = φ1,2 = − (А1е−А2е)/F, (2.4)
де φ1, 2 - контактний потенціал Вольта між металами 1 і 2, В; Aе1, Aе2 - робота виходу електрона з металів 1 і 2, еВ.
Л.І. Антропов, прийнявши в якості другого еталонного металу ртуть, для якої величина роботи виходу електрона з металу і потенціал нульового заряду визначені AеHg = 4,52 еВ, EHgN = -0,19 В, одержав рівняння для розрахунку потенціалу нульового заряду будь-якого металу, якщо для нього відома робота виходу електрона:
ENMe1 = ENMe2 − (A1e − A2e)/F = A1e/F − 4,52 − 0,19 ≈ A1e/F − 4,71 (2.5)
Ця залежність наведена на рис.2.6 для ряду металів, з якої видно, що відхилення дослідних точок від теоретичної прямої в більшості випадків незначні.
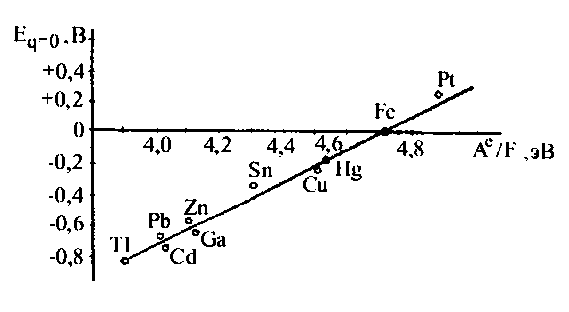
Рисунок 2.6. Зв'язок між потенціалом нульового заряду і роботою виходу електрона у водних розчинах.
Л.І. Антроповим запропонована наведена φ-шкала, в якій за початок відліку прийнята нульова точка. Потенціал φ у наведеній шкалою визначається як різниця між потенціалом електрода EМе в даних умовах і його нульовою точкою:
Φ φ = EМе –EN. (2.6)
Потенціал за φ - шкалою дає характеристику заряду поверхні металу, по якому можна визначити, які поверхнево-активні речовини (катіонні, аніонні або нейтральні) можуть адсорбуватися на поверхні металу в даних умовах. При φ > 0 на поверхні металу переважає адсорбція негативно заряджених часток (аніонів), при φ ˂ 0 - позитивно заряджених часток (катіонів), і при φ ≈ = 0 - молекулярних часток. Адсорбція різних речовин на поверхні металу може дуже сильно змінити швидкість його корозії.
2.5 Термодинаміка корозійних електрохімічних процесів
Основною причиною корозії металів є їх термодинамічна нестійкість. Прагнення металів переходити з металевого стану в іонний (тобто розчинятися) для різних металів є неоднаковим і найбільш точно може бути охарактеризовано зміною вільної енергії при протіканні відповідної реакції окислення в даному середовищі. Відомо, що при самовільному процесі вільна енергія може тільки зменшуватися.
Отже, якщо в даних умовах при переході з металевого стану в іонне спостерігається зменшення вільної енергії, то корозійний процес може протікати самовільно. І навпаки, збільшення вільної енергії в процесі іонізації металу свідчить про неможливість протікання даного процесу самовільно (табл. 2.3).
Таблиця 2.3
Реакція |
Зміна вільної енергії при переході 1 моля металу в іонний стан, кДж / моль |
|
З виділенням водню (РН = 0) |
З поглинанням киcню (рН = 7) |
|
Al →→ Al3+ |
- 160,8 |
- 239,5 |
Zn →→Zn2+ |
- 74,9 |
- 153,7 |
Cr →→ Cr3+ |
- 71,6 |
- 150,3 |
Fe →→ Fe2+ |
- 48,6 |
- 127,3 |
Pd →→ Pd2+ |
+ 95,3 |
+ 16,5 |
Pt →→ Pt2+ |
+ 114,7 |
+ 36,0 |
Au →→ Au3+ |
+ 144,5 |
+ 65,7 |
Як видно з табл. 2.3, паладій, платина, золото є термодинамічно стійкими металами. Решта металів більшою чи меншою мірою мають тенденцію переходити в окислений стан. Термодинамічно стійкі метали в природі, як правило, знаходяться в самородному стані. Це - благородні метали. Всі технічно важливі метали - неблагородні. У природі вони зустрічаються у вигляді руд і солей, тобто в окисленому стані. Термодинамічна стійкість металів дає наближену оцінку корозійної стійкості металів.
При електрохімічної корозії зміна вільної енергії можна виразити таким чином:
ΔG = – EnF, (2.7)
де ΔG - зміна вільної енергії, кДж / моль; Е - ЕРС гальванічної системи, В; n - ступінь окислення металу; F - число Фарадея.
Електрохімічна корозія є можливою, якщо ΔG ˂ 0, тобто зміна вільної енергії має негативне значення, отже, якщо електродний потенціал металу має більш негативне значення в порівнянні з потенціалом деполяризатора.
Принципова можливість протікання процесу електрохімічної корозії металу визначається співвідношенням оборотного потенціалу металу (Еа)утв. та оборотного потенціалу катодного процесу (Ек)утв. в даних умовах:
Ет = (Ек)утв. − (Еа)утв. (2.8)
Для електрохімічного розчинення металу необхідна присутність в електроліті окислювача - деполяризатора, значення оборотного окисно - відновного потенціалу якого має бути більш позитивно, ніж значення оборотного потенціалу металу в даних умовах.
Таким чином, про здатність або нездатність металу до корозії можна судити за величиною його стандартного потенціалу. Однак термодинамічні дані визначають лише можливість протікання корозійного процесу, але не реальну швидкість корозії. Великі від'ємні значення потенціалів не завжди свідчать про високу швидкість корозії (наприклад, для алюмінію і хрому).
2.6 Корозійні гальванічні елементи та електродні реакції
Електрохімічною корозією називається самовільне руйнування металу під дією електричного струму, що виникає внаслідок роботи короткозамкнених гальванічних елементів на поверхні металу за умови її електрохімічної неоднорідності.
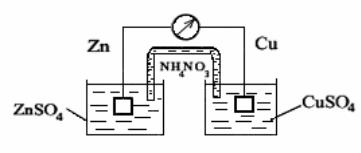
Рисунок 2.7. Схема гальванічного елемента.
Розглянемо роботу звичайного гальванічного елемента, складеного з цинкового та мідного електродів, занурених у розчини солей їх іонів з активністю 1г-іон / л (рис. 2.7):
Zn│ZnSO4 ││CuSO4│Cu.
До замикання електродів на цинку і міді встановлюються рівноважні потенціали:
Zn – 2е ↔ Zn2+, Е0 = – 0,76 В
Сu – 2е ↔ Cu2+, Е0 = + 0,337 В;
При замиканні електродів за рахунок різниці потенціалів в ланцюзі потече струм. Цинк, як більш активний метал, буде розчинятися (Zn - 2е ↔ Zn2+) і посилати електрони в зовнішній ланцюг. На міді буде відбуватися процес приєднання цих електронів катіонами міді з розчину (Сu2+ + 2е ↔ Cu). Ефективність такого гальванічного елемента визначається різницею потенціалів електродів. Електрод (в даному випадку цинковий), що має більш негативний потенціал, називається анодом. Анодний процес - це процес окислення. Електрод (в даному випадку мідний), що має більш позитивний потенціал, називається катодом. Катодний процес - це процес відновлення.
Стандартну ЕРС такого елемента можна розрахувати за значенням енергії Гіббса: E0 = -ΔΔG0Т/2F. За довідником термодинамічних величин, стандартна енергія Гіббса реакції елемента Даніеля - Якобі дорівнює:
ΔΔG0Т = –212,3 кДж/моль = –212,3 кВт·с/моль;
F = 96 500 Кл = 96 500 А·с/моль;
E0 =
![]() = 1,1 В.
= 1,1 В.
Таким чином, причиною появи струму в гальванічному елементі є різниця потенціалів металів. На поверхні будь-якого металу, що знаходиться в електроліті, також виникають короткозамкнені гальванічні елементи, для роботи яких, очевидно, потрібно поділ поверхні на катодні і анодні ділянки, які характеризуються різними значеннями потенціалів. У зв'язку з цим прийнято говорити про електрохімічну гетерогенність, тобто неоднорідность поверхні. Причини виникнення електрохімічної неоднорідності можуть бути різними:
- неоднорідність металевої фази - наявність макро- чи мікро- включення, різна концентрація твердого розчину, неоднакова ступінь обробки поверхні металу і т. д.;
- неоднорідність захисних плівок на поверхні металу - наявність несуцільних окисних плівок, пористість захисних плівок, нерівномірний розподіл на поверхні продуктів корозії (солі і гідроксиди);
- неоднорідність внутрішніх напружень в металі;
- неоднорідність рідкої фази - відмінність в концентраціях власних іонів металу в електроліті, різних солей, іонів водню, кисню і інших окислювачів;
- неоднорідність фізичних умов - різниця температур на ділянках поверхні металу, нерівномірний розподіл зовнішнього електричного поля, неоднакова освітленість поверхні металу та ін.
При роботі корозійного мікрогальванічного елемента (рис. 2.8) одночасно протікають анодний і катодний процеси. Анодний процес - перехід іонів металу в розчин у вигляді гідратованих іонів та утворення некомпенсованих електронів на анодних ділянках:
Me + mН2O = Меn+ · mН2O + ne.
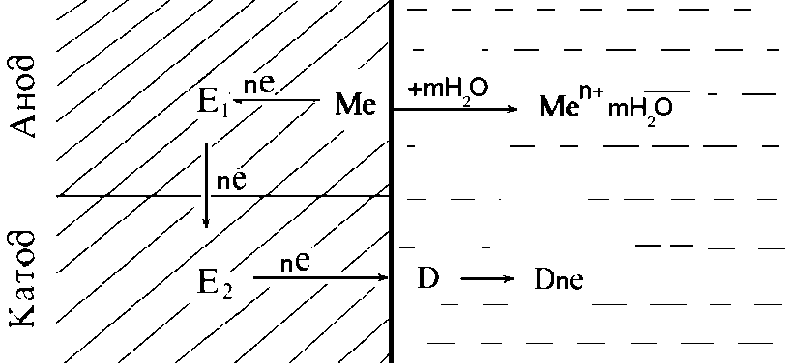
Рисунок 2.8. Схема роботи корозійного мікрогальванічного елемента
Утворені некомпенсовані електрони перетікають по металу від анодних ділянок до катодних. Катодний процес - асиміляція електронів будь-якими іонами або молекулами, що знаходяться в розчині (деполяризатором), і здатними до відновлення на катодних ділянках:
D + nе = [D·ne].
У табл. 2.4 наведені деякі катодні реакції та їх стандартні окисно - відновні потенціали.
Таблиця 2.4
Електродна реакція |
рН |
Е0, В |
H+ + e = 1/2H2 |
0 |
0,000 |
Fe3+ + e = Fe2+ |
0 |
+0,771 |
O2 + 4H+ + 4e = 2H2O |
0 |
+1,229 |
H2O2 + 2H+ + 2e = 2H2O |
0 |
+1,776 |
O2 + 2H+ + 4e = 2OH− |
7 |
+0,815 |
O2 + 2H2O + 4e = 4OH− |
14 |
+0,401 |
Найбільше значення в більшості випадків електрохімічної корозії металів мають наступні катодні реакції:
- киснева деполяризація О2 + 2Н2О + 4е = 4ОН−;
- воднева деполяризації H+ + e = 1/2 H2.
На рис. 2.9 представлена діаграма, що дозволяє визначити можливість протікання корозії з водневою або кисневою деполяризацією.
На діаграмі наведені лінії електрохімічної рівноваги води з продуктами її відновлення (лінія 1) або окислення (лінія 2). Область, яка знаходиться між цими лініями, є областю стійкості води.
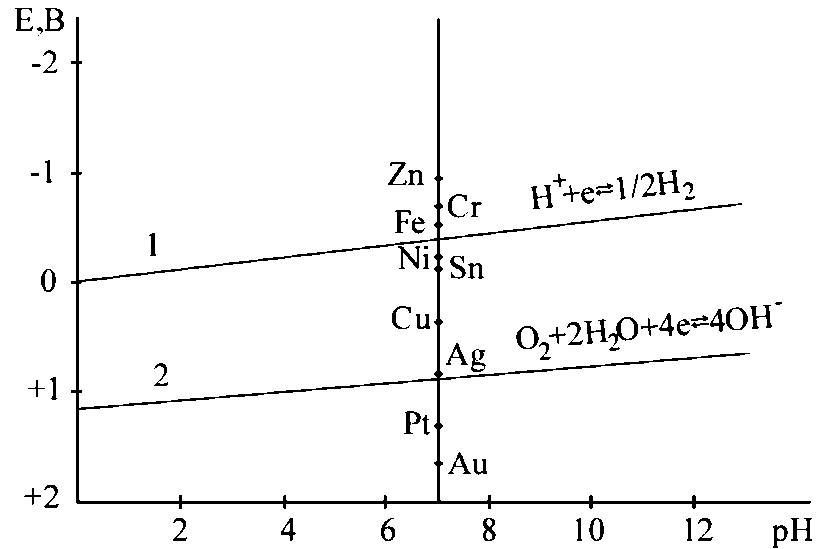
Рисунок 2.9. Діаграма рівноваги водневого і кисневого електродів при різних значеннях рН середовища.
При потенціалах, що лежать поза цією областю, вода термодинамічно нестійка: при потенціалах, що лежать позитивніше лінії 2, вода окислюється, а негативніше лінії 1 - відновлюється. Лінія 1 відповідає рівновазі:
H+ + e = ½H2(у кислому середовищі), (2.9)
H2O + e = ½H2 + OH− (у лужному середовищі). (2.10)
Лінія 2 відповідає рівновазі:
O2 + 4e + 2H2O = 4OH− (2.11)
Виходячи з рівняння (2.9) та враховуючи, що стандартний потенціал водневого електрода прийнято рівним нулю, а негативне значення десяткового логарифма активності іонів водню є величина рН, можна записати:
ЕH+/H2 = − 0,059 pH. (2.12)
З рівняння випливає, що при зміні рН на одиницю потенціал водневого електрода зменшується на 0,059 мВ. Потенціал кисневого електрода позитивніше водневого електрода на 1,23 В, тобто:
ЕOH−/O2 = 1,23 − 0,059pH (2.13)
У кислому середовищі (рН = 0) потенціали водневого і кисневого електродів будуть мати значення:
EH+/H2 = 0; ЕOH−/O2 = + 1,23B.
У нейтральному середовищі (рН = 7) потенціали водневого і кисневого електродів будуть мати значення:
EH+/H2 = − 0,415B; ЕOH−/O2 = + 0,815B.
Таким чином, враховуючи конкретні анодні і катодні реакції, можна визначити можливість протікання корозії і тип катодної деполяризації. На діаграмі відмічені значення стандартних потенціалів деяких металів. Ті метали, потенціали яких розташовуються вище лінії рівноваги водневого або кисневого електроду, можуть кородувати відповідно з водневою або кисневою деполяризацією. Метали, потенціали яких нижче лінії рівноваги кисневого електроду, кородувати не повинні. Вони будуть кородувати тільки в тому випадку, якщо в розчині буде знаходитися будь - який інший деполяризатор, потенціал відновлення якого буде позитивніше потенціалів цих металів.
У реальних умовах, внаслідок гетерогенності поверхні металу, короткозамкнені двохелектродні гальванічні елементи зустрічаються вкрай рідко. Поверхня кородуючого металу представляє собою багатоелектродний, т. б. складаючийся з декількох, відмінних один від одного за потенціалом електродів, гальванічний елемент. Якщо в двоелектродному елементі розподіл катодних і анодних ділянок цілком визначений, то в багатоелектродному елементі цей розподіл залежить від багатьох факторів. Електрод з найбільш негативним потенціалом буде aнодом, а з найбільш позитивним - катодом. Тип електродів з проміжними потенціалами залежить від значення електродних потенціалів з крайніми значеннями, площі електродів, їх поляризуємості і від опору провідників, що з'єднують електроди. Наприклад, в трьохелектродної елементі Zn | розчин | Fe | розчин | Сu цинковий електрод є анодом, мідний - катодом. Тип залізного електрода залежить від його положення. Якщо залізний електрод знаходиться поблизу анода, то він є катодом, якщо ближче до катода - анодом. Якщо один електрод (наприклад катод) має порівняно велику поверхню в порівнянні з іншими електродами, тобто в процесі роботи він мало поляризується, то інші електроди, як правило, перетворюються на електроди протилежного знаку (аноди).
2.7 Діаграма стану системи метал - вода
За термодинамічним даними можна побудувати діаграми стану метал - вода, які називаються діаграмами Пурбе. Ці діаграми дозволяють оцінити можливості протікання процесу корозії металів. Діаграми враховують три можливих типи рівноваги в системі метал - вода:
1. Рівноваги, пов'язані з обміном електричними зарядами:
Ме = Меn+ + ne. (2.14)
Ці рівноваги визначаються тільки величиною потенціалу і не залежать від рН. Лінії, що характеризують ці рівноваги, паралельні осі рН.
2. Рівноваги, не пов'язані з обміном електричними зарядами:
Ме(ОН)n = Меn+ + nОН−. (2.15)
Ці рівноваги визначаються тільки величиною рН і не залежать від потенціалу. Лінії, що характеризують ці рівноваги, паралельні осі потенціалів.
Рівноваги, що залежать як від потенціалу, так і від рН, наприклад на метал-оксидних електродах:
Ме + nОН− = Ме(ОН)n + nе.
Потенціал такого електрода визначається рівнянням:
ЕOH−/Me = E0OH−/ Me − 0,059pH.
З рівняння видно, що лінія рівноваги має нахил, аналогічний нахилу, що характеризує зміну потенціалу водневого електрода. Нахил лінії рівноваги від величини рН в інших випадках визначається, виходячи з рівняння конкретної електродної реакції.
а б
Рисунок 2.10. Діаграми Пурбе для: а – цинку; б – заліза.
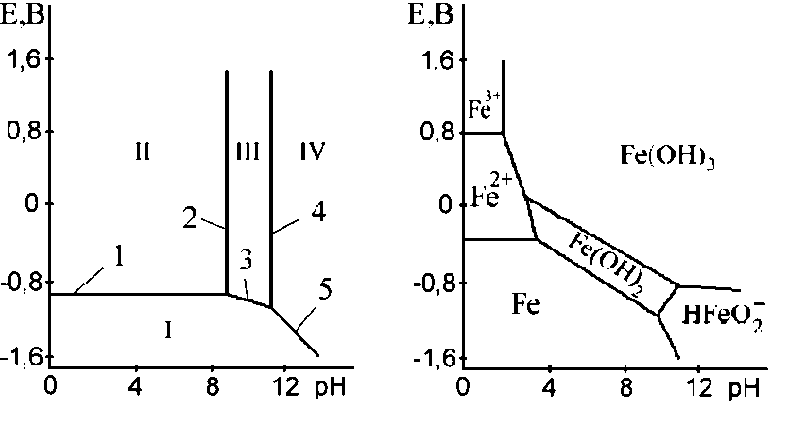
На рис. 2.10 наведені діаграми Пурбе для цинку і заліза. Діаграма для цинку має чотири області: I - область термодинамічної стійкості; II, IV - області корозії і III - область пасивності.
Лінії діаграми відповідають наступним рівноваги:
1 – Zn = Zn2+ + 2e;
2 – Zn2+ + 2OH− = Zn(OH)2;
3 – Zn + 2OH− = Zn(OH)2 + 2e;
4 – Zn(OH)2 + 2OH− = ZnO2 2− + 2H2O;
5 – Zn + 2H2O = ZnO2 2− + 4H+ + 2e.
Таким чином, в області II стійкий іон цинку, в області IV - іон цинкату. Пасивність цинку в області III обумовлена утворенням плівки нерозчинного гідроксиду Zn(OH)2.
На рис. 2.10, б приведена діаграма Пурбе для заліза, на якій також позначені області його термодинамічної стійкості і різні продукти окислення.
На характер корозії впливають різні іони, тому діаграми Пурбе у присутності сторонніх іонів не дають конкретних даних про корозію металу.
.
2.8 Механізм розчинення металів
Першопричиною розчинення металів є їх термодинамічна нестійкість. Мимовільний перехід металів у окислене стан при взаємодії з електролітами може протікати за хімічним і електрохімічним механізмом.
Розчинення металу за хімічним механізмом відбувається в одну стадію на одній і тій же ділянці поверхні металу незалежно від потенціалу без участі вільних електронів, тобто метал вступає з окислювачем в хімічну взаємодію, наприклад, розчинення заліза, хрому і його сплавів в 0,1Н розчині сірчаної кислоти:
Fe + 2H2O → Fe2+ + 2 OH− + H2↑,
2Cr + 6H2O → 2Cr3+ + 6OH− +3H2↑.
або розчинення алюмінію в 50% - вій оцтовій кислоті:
4Al + 3O2 + 12H+ → 4Al3+ + 6H2O.
Розчинення металу за електрохімічним механізмом протікає з участю вільних електронів. При цьому процеси іонізації атомів металу і відновлення окисного компонента корозійного середовища протікають на різних ділянках поверхні металу. Швидкості цих процесів залежать від величин електродних потенціалів ділянок металу. Цей механізм розчинення має місце в більшості випадків корозії металів в електролітах.
Поділ процесу розчинення металу в електроліті на два сполучених процеси - анодний і катодний - полегшує його протікання в порівнянні з хімічною взаємодією. Згідно електрохімічного механізму розчинення металу окислювач є тільки деполяризатором, який забирає валентні електрони металу, але не вступає з ним у хімічну взаємодію. Вторинні процеси утворення продуктів корозії можуть мати місце, але вони не є обов'язковими.
Термодинамічні дані не дозволяють оцінити реальну швидкість електрохімічної корозії, яка визначається конкретними умовами перебігу процесу. Тому необхідно розглянути кінетику цього процесу, що складається з двох сполучно протікаючих реакцій - анодної і катодної. Обидві реакції зв'язані тим, що кількість електронів, які звільнилися при іонізації металу, має бути однаковою з кількістю електронів, взаємодіючих з деполяризатором.
Розглянемо спочатку механізм розчинення чистих металів. Реакцію розчинення металу в чистому вигляді можна записати:
Me → Men+ + ne. (2.16)
При цьому фактичний кінцевий стан катіона в розчині може бути найрізноманітнішим. Це можуть бути гідратовані катіони, а також можливе виникнення зв'язків між катіонами та аніонами, присутніми в розчині. Стадія переносу заряду полягає в переході атомів металу, адсорбованих на самій металевій поверхні і володіють більшою рухливістю в порівнянні з атомами кристалічної решітки, в катіони в безпосередній близькості від поверхні, тому електродну реакцію (2.16) у загальному вигляді можна представити таким чином:
Me(реш) → Me(адс) ; (2.17)
Me(адс) → (Men+)адс + ne; (2.18)
(Men+)адс → (Men+)гидр. (2.19)
Реакція (2.17) являє собою початкову стадію руйнування кристалічної решітки, (2.18) - стадію переносу заряду через подвійний електричний шар, (2.19) - наступну стадію переходу катіонів з подвійного шару в об'єм розчину.
Поверхня реальних полікристалічних твердих металів складається з поверхонь окремих кристалітів і пронизана вузькими перехідними ділянками, де кристалічна структура сильно порушена межами зерен. Крім того, в металі завжди присутні домішки. Місцями з підвищеною вірогідністю переходу атомів металу з решітки на поверхню можуть служити дефекти поверхні металу, що утворюються в результаті механічної обробки, та інші ушкодження. Тому в цілому топографія розподілу активних ділянок поверхні твердого металу, що піддаються розчиненню з найбільшою швидкістю, складна.
Зазвичай найбільш легкий перехід здійснюється з тих місць, де є дефекти структури, "уступи", на яких ослаблено зв'язок деяких атомів з сусідніми атомами по кристалічній решітці. Вважається, що таких активних місць 108-1012 на кожному сантиметрі поверхні, що становить від 10-7 до 10-3 від загальної кількості поверхневих атомів.
2.9 Поляризація електродних процесів
Стандартні електродні потенціали визначають термодинамічну можливість протікання того чи іншого корозійного процесу. У момент замикання ланцюга оборотного гальванічного елемента початкове значення корозійного струму визначається за законом Ома:
I =
![]() ,
(2.20)
,
(2.20)
де Ек(звор) - потенціал катода в розімкнутому стані ланцюга;
Еаа(звор) - потенціал анода в розімкнутому стані ланцюга;
R - загальний опір гальванічного елемента.
Проте в процесі роботи гальванічного елемента початкове значення струму швидко падає, потім встановлюється якесь постійне значення, у багато разів менше початкового. Так як омічний опір у часі істотно не змінюється, то, очевидно, зниження корозійного струму пов'язано із зменшенням початкової різниці потенціалів катода і анода:
I =
![]() ,
(2.21)
,
(2.21)
де Ек і Еа - сталі потенціали катода і анода при даному значенні струму.
Зміна значень потенціалів електродів при проходженні струму називається електродною поляризацією. Сутність явища поляризації зводиться до того, що перехід електронів з анода на катод відбувається швидше, ніж електродні реакції. При анодному процесі швидкість переходу іонів Men+ в розчин відстає від швидкості перетікання електронів. Внаслідок цього у поверхні електрода скупчується надлишок катіонів Men+ і потенціал анода зміщується в позитивну сторону. При катодному процесі на катоді накопичується надлишок електронів, так як катодні деполяризатори не встигають з'єднуватися з електронами, і потенціал катода зсувається в негативну сторону. Явище поляризації в процесах електрохімічної корозії є позитивним,так як воно в сотні разів зменшує швидкості корозії.
Зрушення потенціалу анода в позитивну сторону називають поляризацією анода ΔЕа :
E = Eзвор + ΔEа.
Зрушення потенціалу анода в позитивну сторону може бути викликано декількома причинами: 1) низькою швидкістю анодної реакції корозії металу ne ← neMen+ + mH2O → Men+⋅mH2O, швидкість якої визначається значенням енергії активації. Це призводить до виникнення електрохімічної поляризації, званої перенапруженням іонізації металу ΔЕа(ех); 2) низькою швидкістю дифузії іонів металу з подвійного шару в об'єм електроліту, що призводить до виникнення концентраційної поляризації анода ΔЕа(конц); 3) низькою швидкістю анодної реакції іонізації металу при виникненні його анодної пасивності. Це супроводжується різким гальмуванням швидкості анодного процесу при самовільному падінні щільності струму і значним зміщенням потенціалу анода в позитивну сторону ΔЕа(пас).
Зрушення потенціалу катода в негативну сторону називають поляризацією катода ΔЕк.:
Eк = E0к + ΔEк.
Зрушення потенціалу катода в негативну сторону може бути обумовлений наступними причинами:
низькою швидкістю катодної реакції зв'язування електронів, що надходять з анода: D + ne → [Dne], яка визначається відповідним значенням енергії активації реакції. Це призводить до виникнення перенапруги реакції катодної деполяризації ΔЕк(ек);
2) низькою швидкістю дифузії деполяризатора з об’єму електроліту до катода або продуктів катодної реакції у зворотному напрямку. Це призводить до виникнення концентраційної поляризації катода ΔЕк(конц).
Зменшення різниці початкових електродних потенціалів корозійного елемента внаслідок зсуву потенціалів при протіканні струму призводить до зниження корозійного струму, тобто швидкості корозії. Таке зменшення різниці потенціалів називають поляризацією елемента.
Таким чином, за ступенем зміщення потенціалу електрода при проходженні через нього корозійного струму можна судити про поляризуемість електроду.
Якщо при накладанні струму спостерігається незначне зміщення потенціалу від його початкового значення, то даний електродний процес протікає практично без гальмування. Якщо ж електрод сильно поляризується, це свідчить про ускладнення протікання електродного процесу.
Отже, кінетика електродного процесу якісно може бути визначена залежністю зміни потенціалу електрода при проходженні струму.

а б
Рисунок 2.11. Два види корозійних діаграм.
Поляризаційні криві можуть бути побудовані в різних координатах. На рис. 2.11, а представлена найбільш сувора система координат, де більш позитивні значення потенціалу і струму відкладені відповідно вправо і вгору, а більш негативні значення - вліво і вниз.
Крива А відображає кінетику іонізації металу (анодна крива), крива К - кінетику відновлення деполяризатора (катодна крива). Струм корозії Iкор відповідає потенціалу Ех (компромісний потенціал або потенціал корозії), коли дотримується рівність анодного і катодного струмів Ік = Iа = Iкор.
Діаграма 2.11 б, запропонована Евансом, є найбільш зручною. На цих діаграмах більш негативне значення рівноважного потенціалу відкладається вище, а по осі абсцис відкладають величини і анодного і катодного струмів, незалежно від того, що вони мають протилежне значення. На діаграмі 2.11 б початкова різниця потенціалів Eпоч. = Єкр -Еар представляє собою різницю між початковими (рівноважними) потенціалами анодного і катодного реакцій. При протіканні через корозійну систему струму спостерігаються анодна ΔЕа та катодна ΔЕк поляризації. При цьому встановлюється загальне значення потенціалу Ех. Точка перетину анодної А і катодної К кривих відзначає на осі абсцис величину максимального струму корозії Imax.
Це максимальне значення струму реалізується лише у випадку, коли з опір системи або дорівнює нулю, або мізерно малий. Якщо ж R ≠ 0, швидкість корозії дорівнюватиме не Imax, а дещо меншій величині Iкор . У цих умовах омічне падіння напруги ΔЕом чисельно дорівнює довжині відрізка ab (рис.2.11, б).
Потенціал анода в процесі корозії буде більш негативним на величину ΔЕом потенціалу катода. Таким чином, швидкість корозії залежить від різниці оборотних потенціалів анодної і катодної реакції, поляризуємості електродів і омічного опору корозійного середовища. З наведеної діаграми видно, що струм корозії не може бути розрахований за законом Ома, виходячи з величини початкової різниці потенціалів і опору, що включає опір металу і електроліту, так як. при R → 0, I → ∞.
Для розрахунку швидкості корозії необхідно враховувати кінетичні закономірності катодної і анодної реакцій, що відображається відповідними поляризаційними кривими. Такий розрахунок можна провести, якщо ввести поняття анодної Pа і катодної Pк поляризуємості:
Pа = ΔEа/I; Pк = ΔEк/I.
Ці величини інакше називають поляризаційними опорами, так як вони відображають опір, який чиниться протіканню анодного Ra і катодного Rк процесів. Тому рівняння для розрахунку швидкості корозії можна записати наступним чином:
Ікор =
 (2.22)
(2.22)
Максимальний струм корозії відповідає повній відсутності омічного опору:
Іmax =
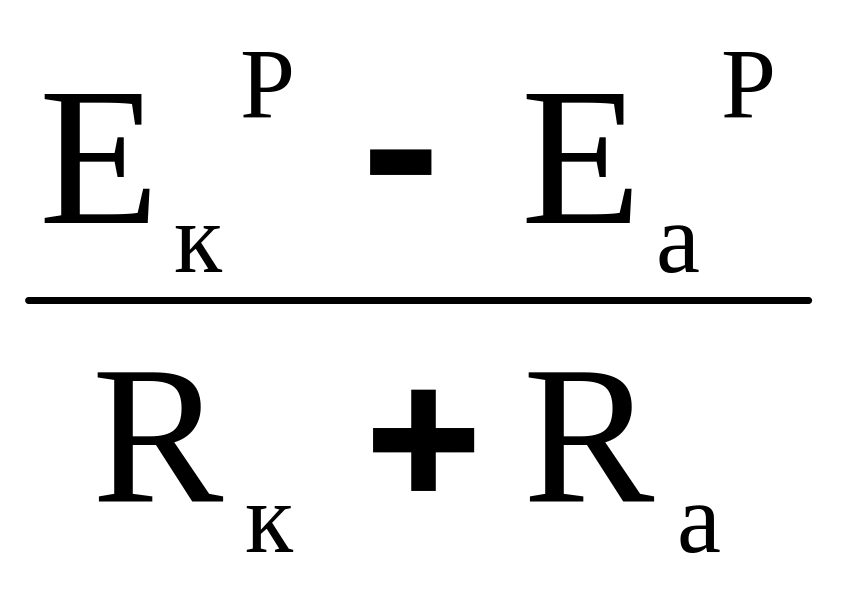 (2.23)
(2.23)
Графічна залежність потенціалу електрода від густини протікає через нього струму i називається поляризаційною кривою (рис. 4.3).
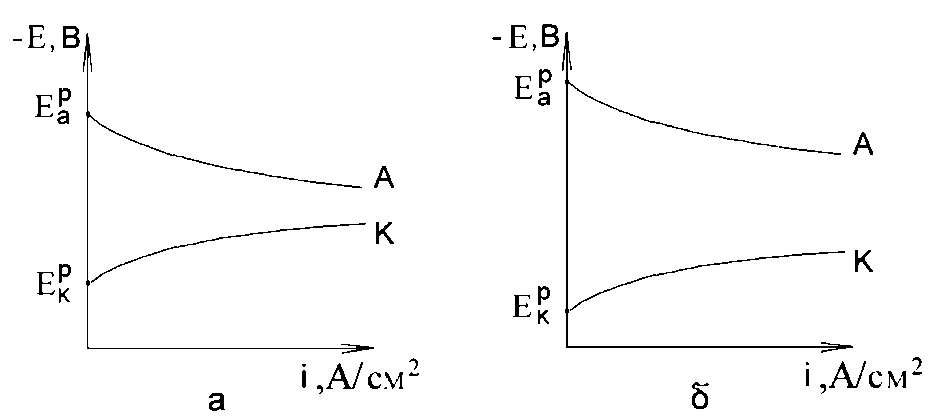
а б
Рисунок 2.12. Поляризаційні криві: а - анодна; б - катодна.
Експериментальна побудова і пояснення поляризаційних кривих корозійних процесів - один з основних методів вивчення механізму електрохімічної корозії. Круте сходження поляризаційних кривих вказує на велику поляризуємість електроду, тобто на загальмованість електродної реакції (рис. 2.12 а). Пологе сходження кривих вказує на малу поляризованість, тобто на безперешкодне протікання катодного і анодного процесів (рис. 2.12 б).
Поляризаційні криві широко використовуються у дослідженнях для пояснення закономірностей корозійних процесів.
2.10 Анодний процес електрохімічної корозії і пасивність металів
Анодний процес електрохімічної корозії металів полягає в іонізації металу:
Ме + mН2О = Меn+∙mН2О + ne.
Можливі й інші реакції анодного окислення металу. У лужному середовищі:
Me + nОН− = МеОnn−(водн) + nН+ + nе.
У кислому середовищі швидкість корозії залежить від рН і від природи аніону кислоти. За даними Я.М. Колотиркіна, анодне розчинення металів йде через утворення комплексу металу з іонами в кілька послідовних стадій:
- специфічна адсорбція аніонів на поверхні металу:
Me + mA− = (МеАm)m−(адс); (2.24)
електрохімічна стадія - перехід комплексу в розчин:
(МеАm)m−(адс) → (МеАm)n−m(водн) + nе; (2.25)
розпад комплексу на прості іони:
(МеАm)n−m = Меn+ (водн) + mА− (водн). (2.26)
Прискорююча дія аніону має місце після досягнення певної концентрації, яка залежить від природи металу і аніону.
У ряді випадків освіта багатозарядної іона металу при його розчиненні відбувається в кілька стадій шляхом послідовного від'єднання електронів:
Me = Me+(водн) + е;
Me+(водн) = Me2+(водн) + е;
Me2+(водн) = Men+(водн) + (n ― 2)е.
Пасивність металу. У ряді випадків при анодній поляризації відбувається різке зниження швидкості розчинення металу. Швидкість корозії багатьох металів різко зменшується при введенні до складу розчину сильних окислювачів. Метал переходить в пасивний стан. Пасивність металу - це стан щодо високої корозійної стійкості, викликаний гальмуванням анодного процесу електрохімічної корозії.
Для ряду металів при анодні поляризації стає можливим не розчинення металу у вигляді катіонів, а утворення на поверхні захисних оксидних плівок. Метали стають пасивними. При виникненні пасивного стану анодний процес розчинення металу гальмується утвореною оксидною плівкою. Потенціал аноду при цьому зміщується в позитивну сторону. Утворення оксидної плівки на поверхні металу різко збільшує загальний опір кола, внаслідок чого зменшується корозійний струм. Явищами пасивності пояснюється мала швидкість корозії нержавіючих сталей, алюмінію,титану та інших металів і сплавів в певних умовах.
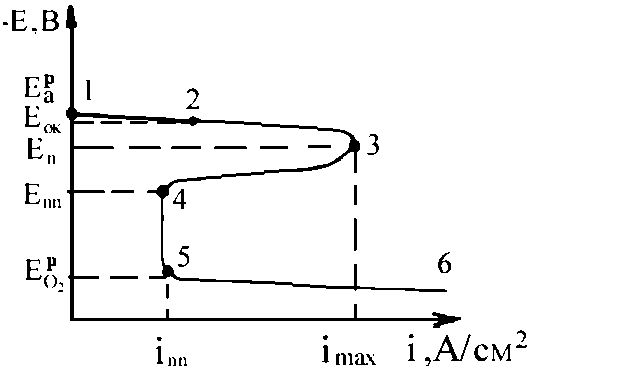
Рисунок 2.13. Анодна поляризаційна крива.
На анодній поляризаційній кривій (рис. 2.13), що відповідає переходу металу в пасивний стан, можна виділити ряд характерних точок і відповідні їм ділянки: ЕaР - початковий потенціал аноду, ЕОК - потенціал освіти оксиду металу, Еп - потенціал початку пасивації, ЕПП - потенціал повної пасивації.
На ділянці 1 - 2 відбувається тільки електрохімічне розчинення металу з утворенням гідратованих іонів. При зсуві потенціалу в позитивну сторону щодо рівноважного потенціалу ЕОК (ділянка 2-3) одночасно з розчиненням металу йде процес утворення оксиду, однак швидкість розчинення металу перевищує швидкість утворення. У точці 3, відповідного потенціалу початку пасивації Еп, швидкості розчинення металу і утворення пасивної оксидної плівки стають рівними, а відповідна цьому значенню потенціалу щільність струму imax (граничний струм пасивації) характеризує максимальну швидкість розчинення металу. Наступний зсув потенціалу в позитивну область (ділянка 3 - 4) обумовлено переважним протіканням реакції утворення пасивної плівки. Все більша частина поверхні металу покривається оксидною плівкою. Омічний опір на межі метал - розчин зростає, а струм поляризації, внаслідок цього зменшується. Точка 4 відповідає завершенню пасивації металу. Потенціал, який відповідає цьому стану, називається потенціалом повної пасивації ЕПП або фладе - потенціалом.
На ділянці 4 - 5 метал знаходиться в пасивному стані. Ця ділянка незалежності струму від потенціалу практично спостерігається до тих пір, поки не буде досягнутий потенціал будь-якої нової анодної реакції, наприклад реакції виділення кисню:
4ОН− − 4е = О2 + 2Н2О.
Новому анодному процесу відповідає часткове або повне розчинення пасивної плівки (ділянка 5 - 6). За ступенем зменшення здатності до пасивації в нейтральних середовищах метали можуть бути розташовані в наступний ряд:
Ti Al Cr Mo Ni Co Fe Mn Zn Cd Sn Pb Cu
Цей ряд характеризує не загальну корозійну стійкість, а тільки ступінь підвищення корозійної стійкості, яка визначається виникненням пасивного стану.
Існують дві основні теоретичні концепції, що пояснюють пасивність металу, - плівкова і адсорбційна. Згідно плівковою теорії (Кістяковський В.А., Акімов Г.В., Еванс), пасивність настає в результаті утворення на поверхні металу фазової плівки товщиною в кілька десятків нанометрів. Ця плівка може представляти собою шар оксиду або гідроксиду. Така плівка збільшується по товщині із зростанням потенціалу у всій області пасивності. Плівкова теорія, заснована на тому, що кисень хімічно зв'язаний з поверхнею металу, не може пояснити всі відомі факти в області пасивності. У багатьох випадках на поверхні металу відсутня фазова плівка.
Згідно адсорбційної теорії (Фрумкін О.М., Колотиркін Я.М., Кабанов В.П.) пасивність настає в результаті адсорбції кисню на поверхні металу. При цьому встановлено, що пасивність може наступити навіть тоді, коли поверхня металу не повністю покрита шаром товщиною в одну молекулу. Цей факт пояснюється тим, що на поверхні металу є обмежене число активних місць і адсорбція кисню на цих місцях («уступах») різко знижує швидкість розчинення металу. Адсорбовані молекули кисню як би «замикають» уступи, тим самим блокуючи процес іонізації в цих найбільш активних місцях. Молекули кисню, що викликають пасивацію металу, утворюються з молекул води або іонів гідроксиду первинно адсорбуються на поверхні металу.
Процес пасивації схематично можна записати так:
Me + 2OH− → MeO + H2O + 2e;
Me + H2O → MeO + 2H+ + 2e,
або Me + 2OH− → Me | O + H2O + 2e;
Me + H2O → Me | O + 2H+ + 2e.
Тут символом Me | O позначена поверхня, закрита шаром хемосорбованого кисню. З рівнянь випливає, що Еп повинен відповідати рівноважному стану обох реакцій і, отже, залежати від рН розчину. Вважаючи, що aMe = aMeO = aH2O = 1, отримаємо:
Еп = Е0 − RT/2F∙ln a2OH − (2.27)
Вразимо a OH- через іонний добуток води KW = a OH-∙∙ aH-, тоді при температурі 25 °С:
Eп = const − 0,059 pH. (2.28)
Звідси випливає, що підвищення рН розчину має зрушувати потенціал пасивації в бік більш негативних значень, тобто полегшувати перехід металу в пасивний стан.
У деяких випадках для пояснення пасивності металів необхідно поєднувати дві основні теорії пасивності - плівкову і адсорбційну, які доповнюють одна одну. Була запропонована гіпотеза плівковий-адсорбційної природи пасивності нержавіючих сталей, згідно якої на поверхні нержавіючих сталей є тонка і щільна захисна плівка. Але під плівкою і в порах знаходяться атоми чи іони кисню або інші окислювачі, хемосорбовані металами, тому поверхня набуває потенціал, близький до окисно-відновного потенціалу корозійного середовища, а активні ділянки плівки (пори) стають анодами. В результаті самополяризації поверхні нержавіючої сталі кисень або комплекси окислювача впроваджуються в метал на анодних ділянках і служать перехідним шаром від металу до захисної плівці, покращуючи їх зчеплення і переводячи метал у пасивний стан.
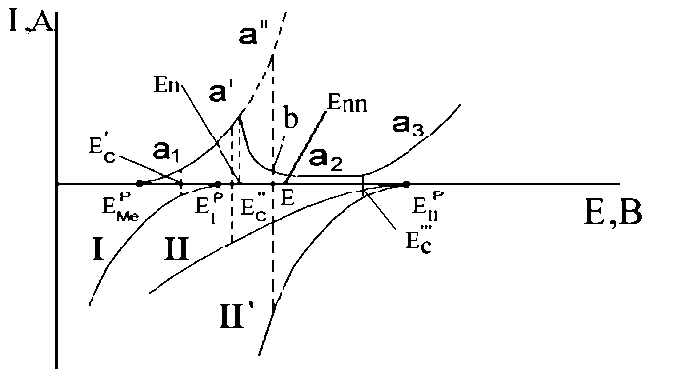
Рисунок 2.14. Поляризаційна діаграма, що показує вплив рівноважного потенціалу окислювача і кінетики його відновлення на можливість пасивації.
Переведення металу в пасивний стан можна здійснити не тільки анодною поляризацією, але і за допомогою окислювачів. Перехід металу в пасивний стан під дією окислювача пов'язаний зі значенням потенціалу, який метал набуває в даному середовищі.
Розглянемо вплив сили окислювача і кінетики відновлення його на можливість пасивації (рис. 2.14). Метал має рівноважний потенціал Еp Ме і анодний поляризаційну криву а1, а2, а3. Окислювач має рівноважний потенціал Ер I, кінетика відновлення якого зображена кривою I.
У цьому випадку встановиться стаціонарний потенціал Ес, що лежить в області активного розчинення. Якщо взяти більш сильний окислювач, рівноважний потенціал якого Eр II, а кінетика відновлення виражена кривою II, то встановиться потенціал Ес ``, теж лежить в області активного розчинення металу. Швидкість окислення металу в цих умовах стане більше. Якщо окислювач II буде відновлюватися з меншим перенапруженням, а катодна крива його піде крутіше (II `), то міг би встановитися потенціал Е, що відповідає рівності швидкостей окислення металу і відновлення окислювача. Але цей потенціал міг би втриматися тільки в тому випадку, якщо б анодна крива в області активного розчинення продовжувалася б вище потенціалу і струму пасивації (пунктир а `, a ``). Однак такого продовження у анодної кривої немає. При потенціалі Е швидкість окислення металу повинна відповідати точці b і бути менше швидкості відновлення окислювача. Тому потенціал Е не може бути стаціонарним. Окислювач зміщує потенціал металу вище значення Еп. Для досягнення стаціонарності потенціал повинен далі зрушуватися в позитивну сторону, поки він не прийме значення Ес `` `, що відповідає рівності швидкостей окислення металу і відновлення окислювача. При цьому метал опиниться в пасивному стані, так як Ес `` `лежить в області ΔЕп.
Таким чином, кінетика відновлення окислювача має велике значення. Будь-який окислювач, рівноважний потенціал якого лежить в області ΔЕп, може викликати пасивацію, якщо перенапруження відновлення його є досить малим. Але більш сильні окислювачі, що відновлюються з великим перенапруженням, можуть утримувати метал у пасивному стані.
Якщо стаціонарний потенціал встановиться в області між Еп і ЕПП на низхідній гілці анодної кривої, то метал опиниться в умовах неповної пасивації і захисний бар'єр буде недосконалий, що може привести до нерівномірної корозії.
Наприклад, сірчана кислота помірної концентрації, в якій окислювачем служить катіон Н+, не пасивує сплави заліза з хромом, хоча вони схильні до пасивації. Висока водневе перенапруження не дозволяє таким сплавам досягти Еп, тому вони швидко піддаються корозії. Для зниження водневого перенапруження сплав легують невеликими кількостями металів, на яких перенапруження є невеликим (Pd, Pt, Cu). Внаслідок зниження перенапруги водню сплави пасивуються і їх швидкість корозії сильно сповільнюється. На можливість пасивації впливають не тільки сила і кінетика відновлення окислювача, а й характер анодної кривої для кородуючого металу. Наприклад, два метали з різними потенціалами пасивації і різним ходом анодних кривих, піддані дії одного і того ж окислювача з однаковим перенапруженням відновлення, можуть опинитися в неоднаковому стані: один в активному, інший в пасивному або один в пасивному, інший в транспасивному.
При зміні зовнішніх умов пасивний метал може знову перейти в активний стан. Цей процес називають активацією або депасивацією. Речовини або процеси, що порушують пасивний стан металів або ускладнюють настання пасивності, називають активаторами або депасиваторами. Активаторами є відновники (Na2SO3, Na2S2O3, Н2 та ін.), катодна поляризація, тобто відновлення поверхні металу постійним електричним струмом від зовнішнього джерела струму, деякі іони (С1–, Br–, S2–, SO42– та ін.), підвищення температури, механічне порушення пасивної плівки.
2.11 Катодний процес електрохімічної корозії металів
При електрохімічній корозії можуть проходити наступні реакції катодної деполяризації:
- розряд катіонів:
H+ + е = l/2H2, Cu2+ + е = Cu+ та ін.
іонізація нейтральних молекул:
О2 + 2Н2О + 4е = 4ОН−, Cl2 + 2е = 2С1−та ін.
- відновлення нерозчинних плівок:
Fе3O4 + Н2О + 2е = 3FеО + 2OН−.
Корозія металевих конструкцій в нейтральних середовищах, морській воді, ґрунті, атмосферних умовах протікає саме внаслідок катодної реакції іонізації кисню. Процес корозії з участю як катодного деполяризатора молекулярного кисню називають корозією з кисневою деполяризацією.
Велике значення для практики має інший випадок катодного деполяризації - розряд катіонів водню. Процес корозії з участю як деполяризатора іонів водню отримав назву корозії з водневою деполяризацією. При корозії всіх металів у кислотах, а магнію і його сплавів навіть у нейтральних середовищах, катодним процесом є розряд катіонів водню.
Корозійні процеси з водневою деполяризацією.
Термодинамічна можливість корозії з водневою деполяризацією визначається співвідношенням:
(Е Ме)обр. < (Е Н2)утв.
де (Е Н2)утв. - оборотний потенціал водневого електрода в даних умовах,
(Е Н2)утв. = (RT / F)2,303lg(aH+ / PH21/2 ) (2.29)
Корозія металів з водневою деполяризацією в більшості випадків має місце в електролітах, що стикаються з атмосферою, парціальний тиск водню в якій Р = 5∙10–2 Па. При визначенні термодинамічної можливості протікання корозійних процесів з водневою деполяризацією оборотний потенціал водневого електрода в цих електролітах слід розглядати, враховуючи реальний парціальний тиск водню в повітрі. У табл. 2.5 наведені значення оборотного потенціалу водневого електрода при температурі 25 °С і різних значеннях рН середовища і парціального тиску водню.
Таблиця 2.5
Р, МПа |
(Е Н2)утв., В |
||
рН 0 |
рН 7 |
рН 14 |
|
5·10−8 |
+ 0,186 |
‒ 0,228 |
‒ 0,641 |
0,1 |
0,00 |
‒ 0,414 |
‒ 0,828 |
Корозія з водневою деполяризацією має місце при досить високій активності водневих іонів у розчині і при досить негативних значеннях потенціалу металу. Реакція розряду іонів водню на катоді виражається рівнянням:
2H+ + 2e = H2.
Цю реакцію можна уявити такою, що протікає в кілька послідовних, пов'язаних між собою стадій: 1) дифузія катіонів водню до поверхні катода, 2) розряд іонів водню з утворенням адсорбованих атомів водню H+ + e = Hадс; 3) рекомбінація або молізація атомів водню з подальшим виділенням у вигляді бульбашок газу Hадс+ + Hадс+ = H2.
Вважають, що на деяких металах третя стадія (рекомбінація атомів) протікає не як хімічний процес, а як процес електрохімічної десорбції, т.б. розряд іона водню відбувається на адсорбованому атомі водню з утворенням молекули:
Hадс + H+ + e = H2 ↑.
Перша стадія - дифузія катіонів водню до поверхні катода - внаслідок великої рухливості іонів водню та його високої концентрації в розчині не є гальмуючою в загальному процесі розряду іонів водню. Найбільш уповільненими стадіями, що визначають швидкість всього процесу, є або стадія електрохімічного розряду іонів водню (друга стадія), або процеси, пов'язані з видаленням адсорбованого водню з поверхні металу (третя стадія).
Роботами А. М.Фрумкіна і його школи встановлено, що для більшості металів найбільш загальмованою стадією є розряд водню. Теорія, що пояснює процес виділення водню гальмуванням електрохімічної стадії розряду H+ + e = Hадс, отримала назву теорії уповільненого розряду. Відповідно до цієї теорії, для того щоб на електроді міг пройти процес розряду іонів водню, необхідна деяка енергія активації. Ця енергія віддається електроду у вигляді надлишкового потенціалу (в порівнянні з рівноважним) при проходженні струму через розчин. Різниця між потенціалом катода при даній щільності струму і рівноважним потенціалом виділення водню в цьому розчині отримала назву перенапруги виділення водню (η H2).
Теорія уповільненого розряду справедлива для металів, погано адсорбуючих водень (цинк, свинець, олово). За теорією сповільненої рекомбінації, уповільненим вважається не розряд іонів водню, а процес молізації, тобто утворення з двох атомів молекули водню. Рекомбінаційна теорія справедлива для металів, що мають високу адсорбційну здатність по відношенню до атомів водню (платина, паладій). Ці метали мають найменші значення перенапруги виділення водню.
Для деяких металів (нікель, залізо, мідь) стадії розряду катіонів водню і рекомбінації протікають з порівнянними швидкостями. Для них перенапруження виділення водню сумарно визначається стадіями уповільненого розряду і видалення водню з поверхні металу. Це метали, добре адсорбуючі водень. Високі значення перенапруги мають метали, що практично не мають здатності до адсорбції водню. Якщо метали розташувати в ряд у порядку зростання їх здатності адсорбувати водень то в цьому ж напрямку зменшується величина перенапруги виділення водню:
Pb Sn Zn Сu Ag Fe Ni W Pd Pt
Встановлено, що перенапруга виділення водню на металах підпорядковується логарифмічній залежності від щільності струму:
ηH2 = а + b lgi, (2.30)
де η H2 - перенапруження, В; i - щільність струму, А/см2, а - коефіцієнт, що залежить від природи металу; чисельно визначається як величина перенапруги при i = 1 А/см2; b - константа, рівна 2,3∙RT / nF.
На рис. 2.15 представлена залежність перенапруги виділення водню від логарифма щільності струму для різних металів. Константа b є тангенсом кута нахилу отриманої прямої до осі абсцис; константа а дорівнює відрізку ординати при щільності струму i = 1 А/см2, тоді lgi = 0. У табл. 2.6 наведено значення перенапруги виділення водню η H2 в залежності від природи металу катода при щільності струму 5 мА/см2.
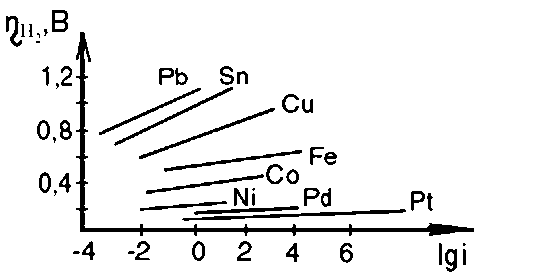
Рисунок. 2.15 Перенапруження виділення водню
Таблиця 2.6
Метал |
Pb |
Zn |
Sn |
Cd |
Cu |
Fe |
Ni |
W |
Pt |
ηH2, В |
1,11 |
0,95 |
0,66 |
0,65 |
0,56 |
0,44 |
0,37 |
0,33 |
0,2 |
Перенапруження виділення водню залежить від природи і складу електроліту. Зі зменшенням кислотності розчину перенапруження виділення водню лінійно зростає, досягаючи максимального значення при рН ≈ 8, потім, із збільшенням лужності розчину, - падає (рис. 2.16).
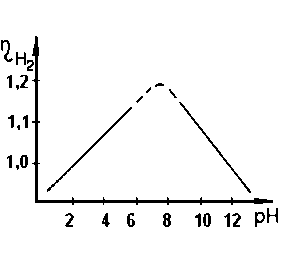
Рисунок 2.16. Залежність перенапруги виділення водню від рН розчину на ртутному катоді при ik = 1∙10–5 А/см2.
В концентрованих розчинах кислот і лугів ця залежність стає складнішою. Зі збільшенням температури електроліту перенапруження виділення водню падає, причому температурний коефіцієнт залежить від природи металу і щільності струму (рис. 2.17). При збільшенні щільності струму температурний коефіцієнт знижується.
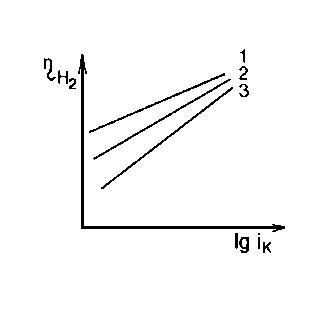
Рисунок 2.17. Залежність перенапруги виділення водню на ртутному катоді від температури і щільності струму: 1 - 200С; 2 - 500С; 3 - 800С.
Таким чином, чим повільніше йде процес виділення водню, тобто чим більше перенапруження виділення водню, тим нижче швидкість електрохімічної корозії.
Корозійні процеси з кисневою деполяризацією
Корозія металів з кисневою деполяризацією є найпоширенішим корозійним процесом. Термодинамічна можливість корозії металів з кисневою деполяризацією визначається рівнянням:
(ЕМе)утв. < (ЕО2)утв.,
де (ЕО2)утв. - оборотний потенціал кисневого електроду в даних умовах,
(Е О2)утв. = (Е О2)0 утв. + (RT / 4F)2,303lg(PО2/aОH–). (2.31)
Корозія металів з кисневою деполяризацією відбувається, в основному, в електролітах, що стикаються з атмосферою, парціальний тиск кисню в якій Р = 0,021 МПа. Отже, при визначенні термодинамічної можливості протікання корозійного процесу з кисневою деполяризацією розрахунок оборотного потенціалу кисневого електроду в цих електролітах слід проводити, враховуючи реальний парціальний тиск кисню в повітрі. У табл. 2.7 наведені оборотні потенціали кисневого електроду при температурі 25 °С, різних значеннях рН середовища і парціального тиску кисню.
Таблиця 2.7
Р, МПа |
(Е О2)утв., В |
||
рН 0 |
рН 7 |
рН 14 |
|
0,021 |
+ 1,218 |
+ 0,805 |
+ 0,381 |
0,1 |
+ 1,229 |
+ 0,815 |
‒ 0,400 |
Реакцію корозії металів з кисневою деполяризацією в загальному вигляді можна записати:
Ме(т) + (n/2)H2O(ж) + (n/4)O2(г) = Ме(OH)n(т).
Корозія протікає, якщо ЕРС (E298)утв. корозійного гальванічного елемента має позитивне значення, а ізобарно - ізотермічний потенціал - від'ємне значення, тобто:
(Е 298) утв. = (ЕО2) утв. ‒ (Е Ме) утв.
Таблиця 2.8
Метал |
Al |
Cr |
Zn |
Fe |
Ni |
Cu |
Продукт корозії |
Al(OH)3 |
Cr(OH)3 |
Zn(OH)2 |
Fe(OH)3 |
Ni(OH)2 |
Cu(OH)2 |
(Е298)утв., В |
+ 2,488 |
+ 1,696 |
+ 1,636 |
+ 1,164 |
+ 1,049 |
+ 0,615 |
ΔG298, кДж/моль |
- 721,5 |
- 491,5 |
- 315,9 |
- 220,4 |
- 204,5 |
- 119,0 |
У табл. 2.8 наведені значення ЕРС (E298)утв. і зміни ізобарно - ізотермічних потенціалів корозійних процесів з кисневою деполяризацією для ряду металів при рН 7, температурі 25 °С і Р = 0,021 МПа.
У водних розчинах електролітів завжди є розчинений кисень, який може виступати катодним деполяризатором. У цьому випадку на катоді відбувається іонізація,тобто відновлення молекулярного кисню:
- в нейтральних і лужних розчинах:
О2 + 2Н2О + 4е = 4OН−,
в кислих розчинах:
О2 + 4Н+ (водн.) +4е = 2Н2О.
Загальний процес кисневої деполяризації можна розділити на дві основні стадії: доставка кисню до катода і електрохімічна реакція іонізації кисню. При корозії з кисневою деполяризацією можливе гальмування загального катодного процесу на першій або другій стадії, або спостерігається сумарне гальмування обох стадій.
При інтенсивному перемішуванні електроліту або за наявності дуже тонкої плівки електроліту на поверхні металу найбільш повільно протікає електролітичне відновлення молекулярного кисню. При повному зануренні металу в спокійний електроліт гальмування катодного процесу пов'язане з утрудненням дифузії кисню до поверхні катода. Утруднення дифузії кисню викликано невеликою концентрацією розчиненого в електроліті кисню (концентраційна поляризація).
Як і при водневій деполяризації, катодний процес іонізації кисню протікає з перенапруженням. Під перенапруженням іонізації кисню розуміють зсув потенціалу катода при даній щільності струму в негативну сторону в порівнянні з рівноважним потенціалом іонізації кисню в тому ж розчині.
Величина перенапруги іонізації кисню підпорядковується логарифмічній залежності від щільності струму, тобто рівнянню Тафеля :
η = a' + b'·lgi,
де b '- константа, пов'язана з механізмом виникнення перенапруги іонізації кисню, і рівна 2,3∙RT / nF; a' - константа, залежна від матеріалу і стану поверхні катода, температури та інших факторів, чисельно обумовлена як величина перенапруги при i = l А / см2.
Аналіз основних катодних процесів електрохімічної корозії показує, що корозія металів з водневою деполяризацією характеризується незначною залежністю швидкості корозії від перемішування розчину, але сильно залежить від значення рН розчину і матеріалу катода.
Для корозії металів з кисневою деполяризацією характерна велика залежність швидкості корозії від перемішування розчину - інтенсивне перемішування в кілька разів збільшує швидкість корозії металів.
2.12 Швидкість електрохімічної корозії
Для розрахунку швидкості електрохімічної корозії використовують такі способи: термодинамічні розрахунки можливості протікання корозійних процесів та визначення рушійної сили процесу; аналітичний та графічний методи розрахунку швидкості процесу;а також показники електрохімічної корозії (глибинний, масовий, об'ємний і струмовий).
2.12.1 Термодинаміка і ЕРС корозійного процесу
Принципова можливість або неможливість самовільного протікання процесу електрохімічної корозії металу визначається зміною ізобарно-ізотермічного потенціалу. Корозійний процес протікає самовільно, якщо ізобарно-ізотермічний потенціал зменшується (ΔG298<0).
Для оцінки можливості самовільного протікання електрохімічної корозії можна користуватися електродними потенціалами. Процес електрохімічної корозії є термодинамічно можливим, якщо дотримується умова:
ΔG298 = – nE298F < 0, (2.32)
де ΔG298 - зміна ізобарно-ізотермічного потенціалу даного корозійного процесу, кДж / моль;
E298 - ЕРС гальванічного елемента, В.
Eутв. = (Eк)утв. – (Eа)утв. , (2.33)
де (Eк) обр - оборотний потенціал катодної реакції в даних умовах, В;
(Eа) обр = (EМе) обр - оборотний потенціал металу в даних умовах, В.
Мимовільне протікання електрохімічного корозійного процесу можливе, якщо:
(Eа)утв. = (EМе)утв. < (Eк)утв.. (2.34)
Різниця потенціалів катодних і анодних реакцій дає кількісне вираження рушійної сили корозійного процесу:
(Eкор)утв. = (Eк)утв. ‒ (EМе)утв.. (2.35)
2.12.2 Контролюючий процес корозії металів
Встановлена швидкість процесу, відповідна силі корозійного струму, визначається гальмуванням протікання струму на окремих етапах.На подолання цього гальмування витрачається початкова різниця потенціалів електродних процесів:
I =
![]() , (2.36)
, (2.36)
де Ра, Рк - поляризуємість анодного і катодного процесів;
Rел – електричний опір розчину.
Поляризуемість анодного і катодного процесів мають розмірність омічного опору, тому їх можна розглядати як опір протіканню анодного і катодного процесів відповідно, або анодниі і катодній поляризації.
(Eк)обр – (Eа)обр = I(Rел + Rа + Rк) = ΔЕел + ΔЕа + ΔЕк, (2.37)
де ΔЕа, ΔЕк - анодна та катодна поляризації;
ΔЕел - падіння напруги в електроліті.
Контролюючим процесом при протіканні корозії називається процес, кінетика якого визначає швидкість корозії, тобто стадія процесу корозії, яка має найбільший опір в порівнянні з іншими, і тому чинить основний вплив на швидкість корозії металу.
Для визначення контролюючої стадії необхідно порівняти величини ΔЕк, ΔЕа і ΔЕел.
Залежно від характеру корозії розрізняються декілька видів контролю електрохімічної корозії металів (рис. 2.18):
- анодний контроль (рис. 2.18, а). Процес протікає при значній анодної поляризації і малою величиною катодного поляризації, тобто ΔЕа >>ΔЕк;
- катодний контроль (рис. 2.18, б).Процес протікає при значній катодного поляризації та малій величині анодної поляризації, тобто ΔЕк > ΔЕа.
- омічний контроль (рис. 2.18, в). Процес не гальмується ні анодної, ні катодного стадіями. Величина корозійного струму визначається омічним опором ланцюга.
- змішаний анодно - катодний контроль (рис. 2.18, г). Процес протікає при значній катодній і анодній поляризаціях, тобто ΔЕк ≈ ΔЕа.
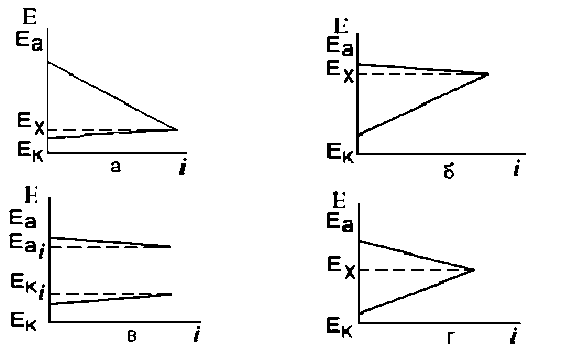
Рисунок 2.18. Основні види поляризаційних корозійних діаграм.
Є й інші види контролю електрохімічної корозії: змішаний катодно - омічний контроль, катодно - анодно - омічний контроль.
2.12.3 Показники електрохімічної корозії металів
Для кількісного вираження середньої швидкості електрохімічної корозії металів найчастіше використовують глибинний, масовий та об'ємний показники корозії.
Основний показник швидкості корозійного руйнування - товщина прокородованого шару металу, мм / рік:
Кν = 8,76![]() , (2.38)
, (2.38)
де q1 - маса металу до корозії, г; q2 - маса металу після корозії, г; ρ - щільність металу, г/см3; S - поверхня металу, м2; τ - час корозії, год.
При рівномірній корозії її швидкість визначають за масою прокородованого металу; г/м2⋅год:
Кm =
![]() .
(2.39)
.
(2.39)
(3.8)
Між глибинним Кν і масовим Кm показниками корозії існує наступна зв'язок: Kv = 8,76 Кm / ρ.
При корозії металу в кислотах швидкість корозії металу може бути визначена за кількістю виділеного водню; см3/см2⋅год:
Коб. =
![]() , (2.40)
, (2.40)
де P - зовнішнє атмосферний тиск при проведенні випробувань, мм рт. ст.; PH2O - тиск насиченої водяної пари при температурі вимірювання, мм рт. ст.; t - температура вимірювання, °С; V - обсяг виділився водню, см3; τ - час корозії, год.
Швидкість електрохімічної корозії можна виразити також через щільність корозійного струму або струмовий показник корозії,тому що за законом Фарадея маса прокорродіровавшего металу пропорційна величині корозійного струму:
Δm = k∙I∙τ, (2.41)
де Δm – втрата маси металу; k – електрохімічний еквівалент; I – сила корозійного струму; τ – час корозії.
Тоді струмовий показник корозії дорівнює:
I = I/Sa, (2.42)
де Sa - площа кородуючого металу.
При рівномірній корозії металів Sa = Sме, тобто загальній поверхні кородуючого металу. У цьому випадку розрахунок швидкості електрохімічної корозії зводиться до визначення величини корозійного струму.
3 Вплив різних факторів на швидкість електрохімічної корозії
Швидкість і характер процесу електрохімічної корозії визначаються внутрішніми, зовнішніми, механічними і конструктивними факторами. Внутрішні фактори електрохімічної корозії пов'язані з природою металу, його структурою, складом, станом поверхні, напругами в металі та ін.
Зовнішні фактори корозії визначаються умовами протікання корозійного процесу, такими, як характер середовища, швидкість її руху, температура розчину та інших
Механічні фактори - це корозійне розтріскування, корозійна втома, корозійне кавітація. Конструктивні фактори визначаються конструктивними особливостями хімічних машин та апаратів.
3.1 Внутрішні фактори корозії
До внутрішніх факторів належать наступні характеристики металу: термодинамічна стійкість, положення в періодичній системі Менделєєва, структура і тип сплаву, наявність домішок, внутрішні напруги та ін.
Для оцінки можливості самовільного руйнування металу необхідно визначити знак зміни ізобарно - ізотермічного потенціалу цього процесу або порівняти значення оборотних потенціалів анодного і катодного процесів.
Термодинамічну стійкість металу можна наближено оцінювати за величиною стандартних електродних потенціалів. Зрушення потенціалу в бік більш позитивних значень можна розглядати як підвищення термодинамічної стійкості металу. Проте теоретична можливість протікання даного корозійного процесу говорить не про реальну швидкість корозії. В якості прикладу розглянемо наступний.
Стандартні значення потенціалів алюмінію і заліза відповідно рівні - 1,67 В і - 0,44 В, т.б. з термодинамічної точки зору алюміній більш схильний до корозії. Однак алюміній стійкий, а залізо не стійке в розведеній сірчаній кислоті, що пов'язано з утворенням пасивної плівки на алюмінії.
Положення металу в періодичній системі однозначно не характеризує його корозійну стійкість, тим не менш щодо корозійної поведінки спостерігаються досить певні закономірності. Найбільш корозійно нестійкі метали знаходяться в головних підгрупах I і II груп. Це лужні і лужноземельні метали. У побічних підгрупах I і II груп корозійна стійкість зростає в міру зростання атомного номера (Cu - Ag - Au, Zn - Cd - Hg). У побічних підгрупах IV та VI груп і в VIII групі знаходяться легко пасивуючі метали, причому зі зростанням атомного номера схильність до пасивації в першому наближенні падає (Ti - Zr - Hf, Cr - Mo - W).
Найбільш корозійностійкі метали знаходяться у восьмому ряду групи VIII (Os, Ir, Pt), а також Au в побічної підгрупі I групи.
Структура металу має різний вплив на швидкість корозії. Так, укрупнення зерна не призводить до збільшення загальної корозії, але сприяє розвитку міжкристалічної.
Металеві сплави по структурі можна розділити на дві групи:
- гетерогенні (двофазні, з включенням надлишкових фаз, композиційні);
- гомогенні (тверді розчини, інтерметаліди, аморфні сплави).
Структура сплаву в значній мірі визначає характер протікання корозійного процесу. Корозійне руйнування сплаву типу «механічна суміш» буде визначатися як атомним (масовим) співвідношенням, так і взаємним розташуванням фаз, що виконують роль катода і анода. Якщо фази розподілені рівномірно і частка анодної складової невелика, то корозія буде суцільною, рівномірною. При нерівномірному розподілі анодної фази корозія буде локальною, вогнища корозії при цьому будуть поширюватися вглиб.
При електрохімічній корозії гетерогенного двофазного сплаву спостерігається найчастіше структурно-виборча корозія, при якій відбувається переважне розчинення електрохімічно більш негативної фази або менш пасивуємої та накопичення на поверхні більш стійкою в корозійному відношенні фази.
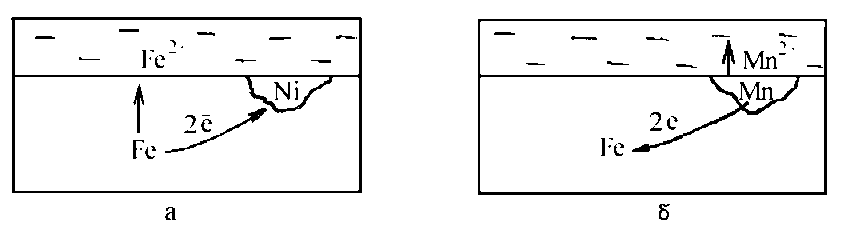
Рисунок 3.1. Схеми корозійних елементів.
Розглянемо два приклади: в одному випадку в залозі знаходиться домішка з більш позитивним електродним потенціалом (рис.3.1, а), в іншому - з більш негативним електродним потенціалом (рис.3.1, б).
Складемо електрохімічну систему для обох випадків:
(−) Fe | розчин | Ni (+); (+) Fe | розчин | Mn (−);
Е0Fe = -0,44 В, Е0Ni = -0,23 В; Е0Fe = -0,44 В, Е0Mn = -1,18 В.
У першому випадку залізо є більш активним, і в корозійному елементі буде грати роль анода, а нікель з більш позитивним потенціалом буде катодом:
(−) Fe → Fe2+ + 2e;
(+) 2H2O + O2 + 4e →→ 4OH− (нейтральне середовище).
У другому випадку залізо з більш позитивним електродним потенціалом буде грати роль катода, а марганець з більш негативним електродним потенціалом - анода:
(−) Mn →→ Mn2+ + 2e;
(+) 2H2O + O2 + 4e →→ 4OH− (нейтральне середовище).
Таким чином, домішки з більш позитивним електродним потенціалом, ніж потенціал основи, будуть прискорювати електрохімічну корозію, а домішки з більш негативним електродним потенціалом - сповільнювати.
У гомогенному сплаві типу «твердий розчин» атоми різних компонентів сплаву не втрачають повністю своєї індивідуальності. Атоми металу, більш корозійностійкого в даних умовах, залишаються такими і в сплаві, і активність атомів сплаву по відношенню до корозійного розчину не вирівнюється. Це пояснюється тим, що при утворенні сплаву типу «твердий розчин» термодинамічна активність атомів сплаву незначно відрізняється від термодинамічної активності атомів в чистому металі.
Вільна енергія атомів при утворенні сплаву зменшується приблизно на 4,8 кДж на 1 г-атом, що відповідає зміні електродного потенціалу металу на 20 мВ.
При утворенні сплаву не відбувається нівелювання спроможності різних атомів до хемосорбції. Так, в сплаві залізо - хром атоми хрому легше утворюють хемосорбційний зв'язок з киснем, легше пасивуються в порівнянні із залізом.
Таким чином, поверхня сплаву типу «твердий розчин» не є гомогенною. У цьому випадку в початковій стадії корозії спостерігається компонентно - виборча корозія. Поверхня поступово збагачується більш електропозитивними включеннями. Причому збагачення поверхні твердого розчину атомами більш стійкого в даних умовах компонента відбувається незалежно від того, чи викликана ця стійкість їхньою термодинамічною стабільністю або більшою схильністю до переходу в пасивний стан. Наприклад, поверхня сплаву цинк - нікель (18% нікелю) в процесі зберігання в корозійному середовищі поступово збагачується атомами нікелю.
Крім субмікронеоднорідностей, що викликаються різнорідністю атомів у сплаві, існує ще енергетична неоднорідність атомів в різних точках кристалічної решітки. Найбільшою енергією володіють атоми, що знаходяться на кутах грані і пов'язані тільки з трьома сусідніми атомами, які і будуть служити центром активного розчинення атомів сплаву.
Ретельність обробки поверхні, шліфування, полірування підвищують стійкість проти корозії. Це пояснюється утворенням на гладкій поверхні більш досконалих, щільних пасивуючих оксидних плівок.
3.2 Зовнішні чинники корозії
До зовнішніх факторів електрохімічної корозії металів відносяться: склад корозійного середовища, її кислотність, температура, швидкість руху рідини, інгібітори та стимулятори корозії та ін.
3.2.1 Вплив кислотності середовища
Вплив концентрації іонів водню в корозійному середовищі на швидкість корозії металів визначається або їх безпосередньою участю в електродному процесі, або їх здатністю впливати на розчинність продуктів корозії, або можливістю утворювати захисні оксидні плівки при зміні рН розчину. Збільшення концентрації іонів водню впливає на швидкість корозії особливо сильно в тому випадку, коли процес корозії контролюється не дифузійними стадіями, а процесом розряду іонів водню.
Швидкість корозії заліза залежить від рН розчину (рис. 3.2, крива 1).
Швидкість корозії |
|
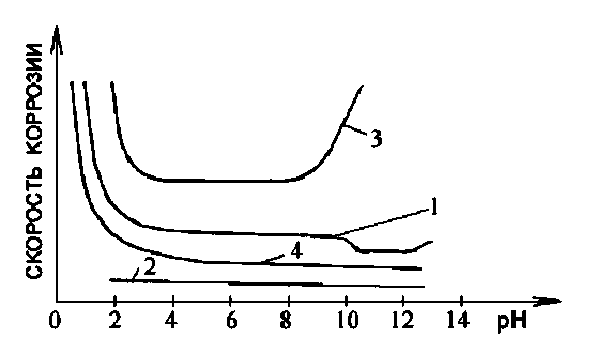
Рисунок 3.2. Вплив рН розчину на характер залежності швидкості корозії для різних металів: 1 - заліза; 2 - благородних; 3 - цинку і алюмінію, 4 - нікелю та кадмію.
В області значень рН від 4 до 10 швидкість корозії не залежить від концентрації водневих іонів. У цьому інтервалі рН швидкість корозії заліза в не перемішуваних електролітах визначається швидкістю дифузії кисню до поверхні металу. У зазначеній області поверхня заліза знаходиться в контакті з лужним розчином, насиченим гідратованих гідроксидом заліза (II), рН якого становить 9,5. У кислому області (рН ˂ 4) плівка гідроксиду заліза розчиняється; катодним процесом є відновлення іонів водню, внаслідок чого відбувається прискорене розчинення заліза. При рН > 10 швидкість корозії знижується в результаті пасивації заліза в лужних розчинах, а потім при рН ˃ 13 настає деяке збільшення швидкості корозії через розчинення пасивної оксидної плівки на залізі в концентрованому лузі. Кожен метал характеризується певною залежністю швидкості корозії від рН розчину (рис. 3.2).
Незалежність швидкості корозії благородних металів (платина, золото, срібло), стійких і в кислих, і в лужних середовищах, від рН виражається прямою 2, що паралельна осі абсцис. Цинк і алюміній нестійкі і в кислотах, і в лугах. У кислотах утворюються катіони Zn2 + і А13 +, а в лугах - аніони ZnO22-і AlO21-, тому на кривій 3 спостерігаються підйоми в кислій і лужній областях. Нікель і кадмій стійкі в нейтральному і лужному розчинах і кородують в кислому (крива 4). Подібні залежності швидкості корозії мають місце при відсутності окислювачів та інших іонів, що утворюють захисні шари на металах.
3.2.2 Вплив складу і концентрації нейтральних розчинів
Корозія більшості металів в нейтральних розчинах протікає з кисневою деполяризацією, і її швидкість сильно залежить від швидкості протікання катодної реакції іонізації кисню і підведення кисню до кородуючої поверхні металу.
Швидкість електрохімічної корозії металів в розчинах солей залежить від природи солі та її концентрації. Водні розчини гідролізуючихся солей впливають на швидкість корозійного процесу збільшенням рН розчину (наприклад карбонат натрію) або зменшенням його (наприклад хлорид амонію). Деякі солі можуть утворювати з первинним катодним чи навіть анодним продуктом корозії металу плівку важкорозчинного з'єднання (наприклад плівки фосфорнокислого заліза на залозі в розчинах фосфорнокислий солі), що призводить до зниження швидкості корозії. Аніони ряду солей руйнують плівку, що призводить до підвищення швидкості корозії.
Якщо в розчині присутні хлориди або сульфати, то швидкість корозії до деякої концентрації солі в розчині зростає, а потім поступово зменшується (рис. 3.3). При введенні у воду невеликих кількостей хлоридів спостерігається збільшення швидкості корозії, що пояснюється активуючою дією іонів хлору на анодний процес. Подальше зниження корозії сталі пояснюється зменшенням розчинності кисню, що є катодним деполяризатором, з підвищенням концентрації солі в розчині.
Іони, що присутні в корозійному середовищі, поділяються на активатори та інгібітори (сповільнювачі) корозії. Іони-активатори бувають аніонного та катіонного типів.
Аніони-активатори (Cl-, Br-, F-) руйнують пасивну плівку або перешкоджають її виникненню, а також полегшують іонізацію металу,пов'язуючи іони металу в комплекси.
Рисунок 3.3. Вплив концентрації солі KCl у розчині на корозію низьковуглецевої сталі.
Катіони-активатори - це іони металів, що мають змінну валентність, наприклад іони дво- і тривалентного заліза. Ці іони, маючи вищу ступінь окислення, беруть участь у катодному процесі, приймаючи електрони:
Mez+ + e → Me(z-1)+,
а маючи нижчу ступінь окислення, можуть взаємодіяти з деполяризатором, наприклад, з киснем:
4Me(z-1)+ + O2+ 4H+ → 4Mez+ + 2H2O.
Розчинність іонів металів в агресивному середовищі значна, тому вони можуть істотно прискорювати катодний процес.
Речовини, додавання яких в розчин у відносно невеликих кількостях призводить до значного зниження швидкості корозії, називаються інгібіторами або сповільнювачами корозії. Сповільнювачі корозії знайшли широке застосування в хімічній промисловості, в системах, що працюють з об'ємом, що мало обновлюємого розчину (системи охолодження, парові котли), при травленні окалини на металах, при межопераційному захисті металевих виробів і їх консервації.
За механізмом дії на електрохімічні процеси інгібітори діляться на анодні, катодні, екрануючі, тобто ізолюючі активну поверхню металу. За умовами застосування їх можна розділити на інгібітори для розчинів і летючі інгібітори для захисту виробів від впливу атмосферної корозії. Механізм захисної дії більшості інгібіторів полягає в їх адсорбції на кородуючій поверхні і наступному гальмуванні катодних або анодних процесів.
До анодних сповільнювачів, в першу чергу, відносяться сповільнювачі окислюючої дії. При цьому на аноді утворюється пасивна плівка, що сприяє зниженню швидкості корозії. Прикладом анодних інгібіторів можуть служити біхромати, хромати, нітрати, нітрити. Нітрати широко застосовуються в якості інгібіторів в парових котлах, нітрити - у машинобудуванні при межопераційному захисту сталевих деталей від корозії.
Поряд з окислювачами гальмувати анодний процес можуть сповільнювачі вторинного дії, що утворюють на поверхні металу пасивні плівки. Плівка утворюється при взаємодії іонів разчиняємого металу із сповільнювачем на кородуючій поверхні металу і гальмує, головним чином, анодний процес. До подібних уповільнювача можна віднести гідроксид і карбонат натрію.
При недостатньому вмісті в розчині анодних інгібіторів вони можуть викликати прискорення корозії. У малих кількостях анодні інгібітори тільки частково пасивують поверхню, але в той же час є ефективними деполяризатором катодного процесу. Внаслідок цього відбувається збільшення швидкості корозії. Тому при використанні анодних інгібіторів необхідно визначити спочатку їх мінімальну захисну концентрацію.
Катодні сповільнювачі зменшують швидкість корозії завдяки зниженню ефективності катодного процесу або зменшення площі катодів. За характером дії вони різні.
Дія катодних сповільнювачів-поглиначів кисню ґрунтується на зменшенні вмісту кисню в розчині і, отже на зниженні швидкості катодного процесу з кисневою деполяризацією. Наприклад:
Na2SO3 +1/2О2 = Na2SО4.
Катодні екрануючі сповільнювачі, що екранують катодні ділянки, утворюють в умовах підлуговування прикатодного простору малорозчинні сполуки:
Са(HCО3)2 + NaOH = СаСОз↓ + NaHCO3 + H2O;
ZnSO4 + 2NaOH = Zn(OH)2↓+ Na2SO4.
Малорозчинні осади карбонату кальцію або гідроксиду цинку, що виділяються на катоді, екранують метал, зменшуючи тим самим ефективність катодного процесу. Катодними сповільнювачами, що підвищують перенапруження катодного процесу, є катіони солей деяких важких металів: хлорид арсену, сульфат вісмуту та ін. Катіони цих солей відновлюються на катоді у вигляді арсену чи вісмуту, внаслідок чого значно підвищується перенапруження виділення водню, тобто дані інгібітори уповільнюють тільки корозію з водневою деполяризацією.
Швидкість руху агресивного середовища в значній мірі визначає корозійне поводження металів. Із збільшенням швидкості руху водопровідної води, яка не містить значних кількостей солей, спочатку відбувається підвищення швидкості корозії заліза і сталі із-за збільшення підведення кисню до мікрокатодів поверхні (рис. 3.4,крива 1).
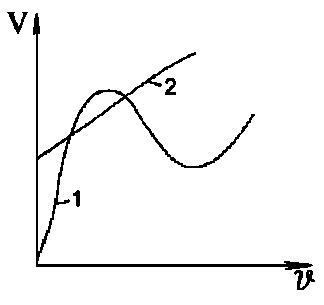
Рисунок 3.4. Залежність швидкості корозії сталі від швидкості руху рідини: 1 - водопровідна вода, 2 - морська вода.
Подальше зниження швидкості корозії при досить швидкому протіканні води пояснюється тим, що велика кількість кисню призводить до пасивації анодних ділянок (кисень виступає як сповільнювач корозії). Нарешті, при дуже великих швидкостях руху води знову спостерігається збільшення швидкості корозії металу внаслідок ерозії, тобто механічного руйнування захисних плівок чи навіть структури самого металу. При наявності в розчині значної кількості активних аніонів пасивація може не настати, тобто відбувається постійне зростання швидкості корозії металу зі збільшенням швидкості руху корозійного середовища, наприклад в морській воді (рис. 3.4, крива 2).
З підвищенням температури швидкість електрохімічної корозії, як правило, зростає. Однак залежність швидкості корозії від температури досить складна, тому що при цьому доводиться враховувати зменшення розчинності кисню з підвищенням температури, зміна структури отримання продуктів корозії, виникнення термогальванічних мікрокорозійних елементів, вплив температури на зміни значень потенціалів для різних металів.
Так як багато хімічних процесів протікають при підвищених тисках, то важливо встановити вплив тиску на швидкість корозії. Підвищення тиску збільшує швидкість корозії металів з кисневою деполяризацією, так як розчинність кисню зростає пропорційно підвищенню тиску в газовій фазі. Швидкість корозії металів з водневою деполяризацією практично не змінюється.
3.2.3 Вплив конструктивних особливостей апаратів на корозійний процес
Конструкція апаратів має істотний вплив на корозійний процес. Застійні зони, концентрації механічних і термічних напруг, контакт різнорідних металів та інші особливості апаратів сприяють електрохімічній корозії. У зв'язку з цим при конструюванні необхідно враховувати наступні моменти:
- контакт різнорідних металів. Чим більше розходження у величині електродних потенціалів контактуємих металів, тим вище корозійний струм. Необхідно підбирати метали, що мають незначну різницю у величині електродних потенціалів, або ізолювати місця їхнього зіткнення;
- чистота обробки поверхні металів. Полірована поверхня менше кородує, ніж грубо оброблена. За наявності ділянок поверхні, що мають різну обробку, можуть виникнути короткозамкнені гальванічні елементи;
- розподіл температури. Велика різниця температур на різних ділянках апарату (наприклад теплообмінника) призводить до утворення термогальванічних елементів, що збільшують електрохімічну гетерогенність кородуючої поверхні. Наслідком цього є підвищення швидкості корозії. Для вирівнювання температури необхідно інтенсивно перемішувати розчини;
- розподіл механічних напружень. При наявності в металі залишкових внутрішніх напружень або доданих ззовні механічних навантажень можуть утворюватися гальванічні елементи на поверхні металу. При цьому на ділянках, що піддаються дії найбільших напружень, з'являються корозійні мікротріщини (розтріскування). Особливо велике напруження виникає у місцях зварювання. Для зменшення напруження необхідно проводити відпал деталей або вузлів апаратів.
- наявність щілин, зазорів і застійних зон. Дуже небезпечними в корозійному відношенні є щілини і зазори, в яких може накопичуватися волога або корозійний розчин, що призводить до сильної місцевої корозії внаслідок нерівномірного аерації (доступу кисню) ділянок поверхні. У застійних зонах значення рН розчину можуть бути зовсім іншими, ніж в об'ємі розчину; в них накопичуються продукти корозії металу, що призводить до збільшення корозії.
3.2.4 Вплив механічних факторів на корозійний процес
Металеві конструкції в процесі експлуатації часто піддаються руйнуванню при одночасному впливі корозійного середовища і механічних напружень. За своїм походженням механічні напруження можуть бути внутрішніми, що виникають в результаті деформації або термообробки металу, або зовнішніми, викликаними прикладеними ззовні навантаженнями (постійними і змінними). Крім того, метал може піддаватися зтираючому або кавітаційному впливу.
Швидкість корозії металевих виробів в агресивних середовищах при одночасному механічному впливі на конструкцію значно вище, ніж при відсутності цього впливу. Залежно від характеру механічної дії розрізняють такі типи корозії:
- корозійне розтріскування - місцеве руйнування металу при одночасному впливі на метал розтягуючих напружень і корозійного середовища;
- корозійна втома - руйнування при одночасному впливі на метал агресивного середовища і знакозмінних напруг;
- корозія при терті - руйнування металу внаслідок механічного зтираючого впливу на метал іншого металу при наявності корозійного середовища або безпосереднього впливу самого рідкого або газоподібного корозійного середовища на метал;
- корозійна кавітація - механічна ударна дія самого агресивного корозійного середовища.
Корозійне розтріскування металів
При накладенні механічних напружень відбувається зниження термодинамічної стійкості металу. На швидкість корозії металів і сплавів в напруженому стані впливають величина механічних напружень, характер катодного процесу, природа аніонів.
У кислих середовищах при додаванні розтягуючих напружень швидкість корозії сталей збільшується. На ступінь збільшення швидкості корозії з водневою деполяризацією впливає природа аніона. Наприклад, у сірчаній кислоті з добавкою хлориду натрію при додаванні розтягуючих напружень швидкість корозії сталі збільшується в більшому ступені, ніж у розчині чистої сірчаної кислоти.
Спостерігаєму закономірність пов'язують з гальмуванням адсорбції поверхнево-активних аніонів на поверхні сталі при додаванні розтягуючих напружень у пружній області. При введенні в розчин сірчаної кислоти аніонів хлору швидкість корозії ненапруженої сталі, за рахунок адсорбції аніонів Cl –, знижується. При застосуванні розтягуючих напружень вигином в пружній області адсорбція аніонів Cl – гальмується, в результаті чого загальна швидкість корозії збільшується.
При корозії з кисневою деполяризацією вплив розтягуючих напружень на швидкість корозії в значній мірі залежить від співвідношення сили корозійного струму і граничного дифузійного струму по кисню. Якщо швидкість корозії напруженого металу лімітується швидкістю дифузії кисню, додавання розтягуючих напружень хоча і полегшує анодний процес, однак не призводить до істотного збільшення швидкості корозії напруженої сталі. Якщо ж швидкість корозії напруженого металу істотно менше граничного дифузійного струму, то додавання розтягуючих напружень викликає збільшення швидкості корозії напруженої сталі.
Постійні розтріскуючі напруження (зовнішні чи внутрішні) збільшують швидкість загальної корозії металу приблизно пропорційно їх величині. При цьому відбувається, головним чином, місцева корозія або корозійне розтріскування. Корозійне розтріскування металів при одночасному впливі агресивного корозійного середовища та розтягуючих напружень характеризується утворенням тріщин у площинах, перпендикулярних до напрямку розтягуючих напружень. До корозійного розтріскування схильні багато металів: лужна крихкість металу парових котлів, розтріскування деяких корозійностійких, наприклад, хромонікелевих, сталей.
Корозійно-механічні тріщини поступово зароджуються на металевій поверхні під впливом локалізації анодного процесу та розтягуючих напружень на окремих її ділянках (наприклад подряпини, дефекти від обробки, дефекти захисної плівки та ін.) Розвиток корозійних тріщин відбувається в результаті дії трьох факторів: електрохімічного - через неоднорідність структури поверхні металу (дефекти захисних плівок є активними анодами); механічного - нерівномірність напружень призводить до електрохімічної неоднорідності; адсорбційного - утворення тріщин у поверхневому шарі металу полегшується під розклинюючим впливом адсорбованих з розчину поверхнево-активними речовинами.

а б в
Рисунок 3.5. Типовий вигляд корозійного розтріскування у воді, що містить 200 мг/л С1 , температура 320°С, збільшення в 900 разів:
а - сталь 03Х16Н4С2, міжкристалітне руйнування, слабо розгалужена тріщина, огинання фериту; б - сталь 03Х12К12Н2, транскристалітне руйнування; в - сталь 03Х12К12Д2, міжкристалітне руйнування, розгалужена тріщина.
При корозійному розтріскуванні металу можна виділити три основні періоди у руйнуванні металу. Інкубаційний період - це процес поступового утворення на поверхні металу мікрокорозійних тріщин під впливом корозійного середовища і локалізації розтягуючих напружень. Найбільш сприятливими умовами для зародження мікротріщин є такі, при яких і розтягуючі напруження, і корозійний процес локалізуються на окремих ділянках поверхні металу. У сильно агресивних середовищах, що викликають рівномірну загальну корозію, ймовірність корозійного розтріскування набагато менше, ніж в середовищах, які викликають місцеве руйнування металу (азотнокислі солі і луги для вуглецевих сталей).
Розвиток корозійної тріщини відбувається при спільній дії корозійного середовища та розтягуючих напружень в металі. Зростання тріщини можна розглядати як безперервний електрохімічний процес, що в значній мірі прискорюється накладеними напруженнями розтягування (рис. 3.5).