

18. В тетради
19. КИРХГОФА УРАВНЕНИЕ, термодинамич. соотношение, определяющее зависимость теплового эффекта реакции от температуры Т. Для реакции при постоянном давлении р кирхгофа уравнение имеет вид:
![]()
где
DH=Qp - тепловой
эффект, равный изменению энтальпии
системы вследствие реакции, DСр -
изменениетеплоемкости системы.
Если реакцию записать в форме где
vi - стехиометрия.
коэф. вещества Ai (vi>0
для продуктов ![]() реакции,
vi<0
для исходных веществ), то DH и DСр рассчитывают
по ф-лам:
реакции,
vi<0
для исходных веществ), то DH и DСр рассчитывают
по ф-лам:
![]()
где Hi - парциальная молярная энтальпия вещества Аi, Срi - его парциальная молярная теплоемкость; тепловой эффект относят к одному пробегу реакции, при котором число молей i-го вещества изменяется на величину vi. Значение DH (DСр) представляет разность между энтальпиями (теплоемкостями) продуктов реакции и исходных веществ, взятых в стехиометрич. соотношении. При расчетах Hi и Срi приравнивают обычно соответствующим значениям для чистого вещества Аi. Для реакции при постоянном объеме кирхгофа уравнение имеет вид:
![]()
где
DU=Qv - тепловой
эффект, равный изменению внутр. энергии
системы при одном пробеге реакции; ![]() разность
теплоемкостей СV продуктов
реакции и исходных веществ. кирхгофа
уравнениепозволяет
рассчитать тепловой эффект реакции при
любой температуре Т2,
если известен тепловой эффект для к.-л.
одной температуры T1 и
имеются данные о зависимости теплоемкостей
участвующих в реакции веществ от
температуры в интервале между Т1 и
Т2.
Интегрируя ур-ние (1), получим:
разность
теплоемкостей СV продуктов
реакции и исходных веществ. кирхгофа
уравнениепозволяет
рассчитать тепловой эффект реакции при
любой температуре Т2,
если известен тепловой эффект для к.-л.
одной температуры T1 и
имеются данные о зависимости теплоемкостей
участвующих в реакции веществ от
температуры в интервале между Т1 и
Т2.
Интегрируя ур-ние (1), получим:
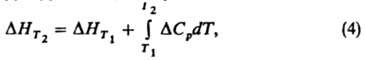
где DHT1 и DHT2 тепловые эффекты реакции при температурах Т1 и Т2. В частности, широко применяется след, форма ур-ния (4):
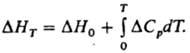
Здесь DH0 - гипотетич. тепловой эффект, который бы наблюдался, если бы принимаемые в расчетах температурные зависимости теплоемкости были справедливы вплоть до абс. нуля температур. Ур-ние (1) применяют для приближенного определения зависимости теплот испарения и сублимации от температуры. В этих случаях DСр=Спарр-С*p (или Српар-Сртв). Однако при точных вычислениях следует учитывать, что с изменением температуры равновесное давление двухфазной системы изменяется. Ур-ния (1) и (2) выведены Г. Р. Кирхгофом в 1858.
20. Энергия химической связи, равна работе, которую необходимо затратить, чтобы разделить молекулу на две части (атомы, группы атомов) и удалить их друг от друга на бесконечное расстояние. Например, если рассматривается Э. х. с. H3C—H в молекуле метана, то такими частицами являются метильная группа CH3 и атом водорода Н, если рассматривается Э. х. с. Н—Н в молекуле водорода, такими частицами являются атомы водорода. Э. х. с. — частный случай энергии связи, обычно ее выражают в кдж/моль (ккал/моль); в зависимости от частиц, образующих химическую связь, характера взаимодействия между ними (ковалентная связь,водородная связь и другие виды химической связи), кратности связи (например, двойные, тройные связи) Э. х. с. имеет величину от 8—10 до 1000 кдж/моль. Для молекулы, содержащей две (или более) одинаковых связей, различают Э. х. с. каждой связи (энергию разрыва связи) и среднюю энергию связи, равную усредненной величине энергии разрыва этих связей. Так, энергия разрыва связи HO—H в молекуле воды, т. е. тепловой эффект реакции H2O = HO + H равен 495 кдж/моль, энергия разрыва связи Н—О в гидроксильной группе — 435 кдж/моль, средняя же Э. х. с. равна 465 кдж/моль. Различие между величинами энергий разрыва и средней Э. х. с. обусловлено тем, что при частичной диссоциации молекулы (разрыве одной связи) изменяется электронная конфигурация и взаимное расположение оставшихся в молекуле атомов, в результате чего изменяется их энергия взаимодействия. Величина Э. х. с. зависит от начальной энергии молекулы, об этом факте иногда говорят как о зависимости Э. х. с. от температуры. Обычно Э. х. с. рассматривают для случаев, когда молекулы находятся в стандартном состоянии или при 0 К. Именно эти значения Э. х. с. приводятся обычно в справочниках. Э. х. с. — важная характеристика, определяющая реакционную способность вещества и использующаяся при термодинамических и кинетических расчетах реакций химических. Э. х. с. может быть косвенно определена по данным калориметрических измерений (см.Термохимия), расчетным способом (см. Квантовая химия), а также с помощью масс-спектроскопии испектрального анализа.
21. Если исходить из первого закона термодинамики, то можно допустить протекание любого процесса, который не противоречит закону сохранения энергии. В частности, при теплообмене можно было бы предположить, что теплота может передаваться как от тела с большей температурой к телу с меньшей температурой, так и наоборот. При этом согласно первому закону термодинамики накладывается только одно условие: чтобы количество теплоты, отданной одним телом, равнялось количеству теплоты, принятой другим телом.
Между тем, из опыта известно, что теплота всегда самопроизвольно передается только от более нагретых тел к менее нагретым. Самопроизвольный или естественный процесс теплообмена обладает свойством направленности в сторону тел с более низкой температурой. Причём он прекращается при достижении равенства температур участвующих в теплообмене тел. Однако, возможен и обратный, не самопроизвольный (или противоестественный) процесс передачи теплоты от менее нагретых тел к более нагретым (например, в холодильных установках), но для осуществления его требуется подвод энергии извне как бы для компенсации протекания процесса.
Констатация этой особенности теплоты, проявляющейся в процессе ее передачи, является одной из сторон сущности второго закона термодинамики, который Р. Клаузиус (1850 г.) сформулировал так: теплота не может сама собой переходить от менее нагретого тела к более нагретому, т. е. некомпенсированный переход теплоты от тела с меньшей температурой невозможен.
Еще одна особенность теплоты наиболее ярко раскрывается при рассмотрении процесса преобразования ее в работу. Опыт показывает, что работа может быть полностью превращена в теплоту (например, посредством трения) без каких-либо дополнительных условий или компенсации. Обратное же превращение теплоты в работу требует дополнительного самопроизвольного процесса или компенсации.
Второй закон термодинамики устанавливает направленность и условия протекания естественных процессов. Так же, как и первый закон термодинамики, он был выведен на основании экспериментальных данных.
Опыт показывает, что превращение теплоты в полезную работу в тепловых двигателях может происходить только при переходе теплоты от нагретого тела к холодному, то есть при наличии разности температур между теплоотдатчиком (нагревателем) и теплоприемником (холодильником). При этом вся теплота не может быть превращена в работу.
Устройство, которое без компенсации полностью превращало бы в работу теплоту какого-либо источника, называется вечным двигателем второго рода.
Таким образом, второй закон термодинамики утверждает, что создание вечного двигателя второго рода невозможно.
Открытие второго закона термодинамики связано с анализом работы тепловых машин. Впервые сущность этого закона изложил в 1824 г. французский инженер С. Карно в работе «Размышление о движущей силе огня и машин, способных развивать эти силы». С. Карно впервые указал на возможность превращения теплоты в полезную работу в двигателях лишь при наличии двух источников теплоты: одного с более высокой температурой (нагреватель с температурой T2) и другого с меньшей температурой (холодильник с температурой T1).
Позднее Р. Клаузиус и В. Томсон (Кельвин) дали наиболее общие формулировки второго закона термодинамики, из которых следует, что:
1. Невозможен процесс, при котором теплота переходила бы самопроизвольно от холодных тел к телам нагретым.
2. Не вся теплота, полученная от теплоотдатчика, может перейти в работу, а только часть ее. Часть теплоты должна перейти в теплоприемник.
22.
23.
24.
25.
26. Свободная энергия Гиббса (или просто энергия Гиббса, или потенциал Гиббса, или термодинамический потенциал в узком смысле) — это величина, показывающая изменение энергии в ходе химической реакции и дающая таким образом ответ на вопрос о принципиальной возможности протекания химической реакции; это термодинамический потенциал следующего вида:
![]()
Энергию Гиббса можно понимать как полную химическую энергию системы (кристалла, жидкости и т. д.)
Понятие энергии Гиббса широко используется в термодинамике и химии.
Самопроизвольное протекание изобарно-изотермического процесса определяется двумя факторами: энтальпийным, связанным с уменьшением энтальпии системы (ΔH), и энтропийным T ΔS, обусловленным увеличением беспорядка в системе вследствие роста ее энтропии. Разность этих термодинамических факторов является функцией состояния системы, называемой изобарно-изотермическим потенциалом или свободной энергией Гиббса (G, кДж)
Классическим определением энергии Гиббса является выражение
где ![]() — внутренняя
энергия,
— внутренняя
энергия, ![]() — давление,
— давление, ![]() — объём,
— объём, ![]() —
абсолютная температура,
—
абсолютная температура, ![]() — энтропия.
— энтропия.
ГЕЛЬМГОЛЬЦА ЭНЕРГИЯ (изохорно-изометрический потенциал - свободная энергия), один из потенциалов термодинамических, обозначаемый F (иногда А) и определяемый разностью между внутренней энергией (U) и произведением термодинамической температуры (Т) на энтропию (S): F = U - TS. Работа системы в равновесном изотермическом процессе равна убыли энергии Гельмгольца; самопроизвольно такой изотермический процессможет протекать только в сторону уменьшения Гельмгольца энергии.
27. (Большинство самопроизвольных процессов -экзотермические процессы,протекающ. с выделением энергии.)В основе протекания процессов лежит принцип минимума энергии,т.е система всегда стремится перецти из сост. с большей энергией в сост с меньшей энергией.2 критерий самопроизвольного протекания процессов явл.увеличениеэнтропии системы,процесс может протекать самопроизвольно ,даже без поглощ. эн.,если при этом увелич.энтропия.
28.
Хими́ческий
потенциа́л ![]() — термодинамическая функция,
применяемая при описании состояния
систем с переменным числом частиц.
Определяет изменение термодинамических
потенциалов (энергии Гиббса, внутренней
энергии, энтальпии и т. д.) при изменении
числа частиц в системе. Представляет
собой энергиюдобавления
одной частицы в систему без совершения
работы. Определение химического
потенциала можно записать в виде:
— термодинамическая функция,
применяемая при описании состояния
систем с переменным числом частиц.
Определяет изменение термодинамических
потенциалов (энергии Гиббса, внутренней
энергии, энтальпии и т. д.) при изменении
числа частиц в системе. Представляет
собой энергиюдобавления
одной частицы в систему без совершения
работы. Определение химического
потенциала можно записать в виде:
![]()
где Е — энергия системы, S — её энтропия, N — количество частиц в системе.
Эта формула определяет, кроме химического потенциала , также давление P и температуру T.
Для систем, состоящих из одного компонента, можно доказать, что химический потенциал задаётся формулой
![]() ,
,
где ![]() — потенциал
Гиббса.
— потенциал
Гиббса.
29.
30. Если реакция протекает обратимо, то ΔG = 0.
Если
реакция протекает необратимо, то ΔG ≤
0 и можно рассчитать изменение ΔG. 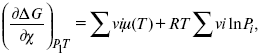
где χ – пробег реакции – величина, которая показывает, сколько молей изменилось в ходе реакции. I сп – характеризует равновесное и неравновесное состояние реакции, II сп – характеризует только неравновесные состояния.
Если
д χ = 1,
то 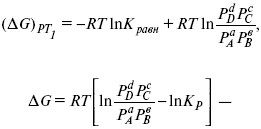
это уравнение изотермы химической реакции
. 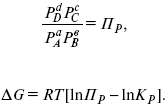
31.
32.
33. Факторы влияющие на химическое равновесие:
1) температура
При увеличении температуры химическое равновесие смещается в сторону эндотермической (поглощение) реакции, а при понижении в сторону экзотермической (выделение) реакции.
CaCO3=CaO+CO2 -Q t↑ →, t↓ ←
N2+3H2↔2NH3 +Q t↑ ←, t↓ →
2) давление
При увеличении давления химическое равновесие смещается в сторону меньшего объёма веществ, а при понижении в сторону большего объёма. Этот принцип действует только на газы, т.е. если в реакции участвуют твердые вещества, то они в расчет не берутся.
CaCO3=CaO+CO2 P↑ ←, P↓ →
1моль=1моль+1моль
3) концентрация исходных веществ и продуктов реакции
При увеличении концентрации одного из исходных веществ химическое равновесие смещается в сторону продуктов реакции, а при увеличении концентрации продуктов реакции-в сторону исходных веществ.
S2+2O2=2SO2 [S],[O]↑ →, [SO2]↑ ←
Катализаторы не влияют на смещение химического равновесия!
34.
35.
36. закономерности, которые проявляются в примерах нарушения химического равновесия, представляют собой частные случаи общего принципа, определяющего влияние различных факторов на равновесные системы. Этот принцип, известный под названием принципа Ле Шателье, в применении к химическим равновесиям можно сформулировать так:
Если на систему, находящуюся в равновесии, оказать какое-либо воздействие, то в результате протекающих в ней процессов равновесие сместится в таком направлении, что оказанное воздействие уменьшится.
37.
38. Молекулярность реакции - это число молекул исходных веществ, принимающих участие в одном (единичном) химическом превращении. При этом число молекул образующихся продуктов не имеет значения.
В соответствии с приведенным определением различают реакции:
1) мономолекулярные, в которых только один вид молекул участвует в превращении, причем стехиометрический коэффициент в уравнении равен единице, например,
запись А → С означает, что молекула вещества А превращается в молекулу вещества С;
2) бимолекулярные, в которых участвуют два различных вида молекул или две молекулы одного вида (стехиометрический коэффициент во втором случае равен двум), например, А + В→С или 2А → С;
3) тримолекулярные, в которых участвуют три молекулы одного или разного видов, например, А + В + D → С или
2 А + В → С, или 3А→С.
Реакции более высокой молекулярности маловероятны. Связано это с причиной, о которой говорилось ранее.
Выше было сказано, что порядок химической реакции выражается
суммой:
где аi - показатели степени концентрации исходных веществ в уравнении действующих масс.
Они приравнивались стехиометрическим коэффициентам компонентов химической реакции. Исходя из этого можно сделать заключение, что молекулярность и порядок реакции это одинаковые величины. Однако, это не всегда так. Порядок реакции или равен молекулярности или, в большинстве случаев, меньше её. Расхождение между порядком реакции и её молекулярностью может быть вызвано разными причинами.
1. Молекулярность реакции величина теоретическая, а порядок реакции - экспериментальная. Между теоретическими и экспериментальными величинами почти всегда есть различия.
2. Если, например, в реакции
bB + dD = P,
скорость которой W = КСBbCD d
один из компонентов, например, компонент B, находится в избытке, то в ходе данной реакции его концентрация будет изменяться незначительно и в уравнении скорости реакции можно принять СB = const. Но в таком случае скорость реакции практически зависит от концентрации только компонента D, то есть W = К1CD d тогда порядок реакции равен d, а молекулярность реакции (b + d).
3. Если данная реакция является гетерогенной, то в зависимости от условий протекания порядок такой реакции может быть различным.
4. Порядок каталитической реакции также может отличаться от молекулярности, причина - сложный механизм таких реакций.
5. Для сложной реакции, протекающей в несколько стадий, порядок реакции и её молекулярность не совпадают. В данном случае порядок реакции определяет какая-либо промежуточная (лимитирующая) стадия. Как правило порядок этой стадии отличается от молекулярности сложной реакции.
39.
Константа скорости реакции есть функция от температуры; повышение температуры, как правило, увеличивает константу скорости. Первая попытка учесть влияние температуры была сделана Я. Г. Вант-Гоффом, который сформулировал следующее эмпирическое правило:
При повышении температуры на каждые 10 градусов константа скорости элементарной химической реакции увеличивается в 2 – 4 раза.
Величина, показывающая, во сколько раз увеличивается константа скорости при повышении температуры на 10 градусов, есть температурный коэффициент константы скорости реакции γ. Математически правило Вант-Гоффа можно записать следующим образом:
![]() (II.29)
(II.29)
![]() (II.30)
(II.30)
Однако правило Вант-Гоффа применимо лишь в узком температурном интервале, поскольку температурный коэффициент скорости реакции γ сам является функцией от температуры; при очень высоких и очень низких температурах γ становится равным единице (т.е. скорость химической реакции перестает зависеть от температуры).
40.