

3
|
|
ЗМІСТ |
|
|
|
Зміст …………………………………………....…………………...………… |
3 |
||||
Загальні правила безпеки при виконанні робіт у мікробіологічній |
|
||||
лабораторії …………….....................................………………..………...... |
4 |
||||
Організація занять ……………………………………………….………….. |
9 |
||||
Лабораторне заняття 1. Загальні правила роботи |
з |
бактеріями і |
|
||
|
бактеріофагами …………………….……..……... |
10 |
|||
Лабораторне заняття 2. Цитоплазматична спадковість у дріжджів ......... |
31 |
||||
Лабораторне заняття 3. Одержання |
гібридів |
і |
принципи |
|
|
|
гібридологічного аналізу ………………….…… |
40 |
|||
Лабораторне заняття 4. Мутагенез у мікроорганізмів ………….……….. |
44 |
||||
Лабораторне заняття 5. |
Відбір та ідентифікація біохімічних мутантів ….. |
54 |
|||
Лабораторне заняття 6. |
Флуктуаційний тест Лурія й Дельбрюка ………. |
59 |
|||
Лабораторне заняття 7. |
Генетичне картування прокаріот ………….……. |
64 |
|||
Лабораторне заняття 8. |
Методи виділення ДНК із клітин |
|
|||
|
мікроорганізмів …………………………….…….. |
70 |
|||
РЕКОМЕНДОВАНА ЛІТЕРАТУРА ………………..…….………………… |
78 |
||||

4
ЗАГАЛЬНІ ПРАВИЛА БЕЗПЕКИ ПРИ ВИКОНАННІ РОБІТ У МІКРОБІОЛОГІЧНІЙ ЛАБОРАТОРІЇ
При роботі у мікробіологічній лабораторії слід суворо дотримуватись вимог, викладених у інструкції з техніки безпеки. У разі, якщо студент не ознайомлений з зазначеними вимогами, він повинен повідомити про це викладача. Студент несе персональну відповідальність за власну безпеку під час перебування у лабораторії, що підтверджує підписом у журналі з техніки безпеки при проведенні інструктажу.
Перед початком кожної лабораторної роботи студент повинен ознайомитись з особливостями техніки безпеки при проведенні лабораторної роботи та поставити свій підпис у журналі.
До роботи не допускаються студенти, які мають пошкодження на відкритих ділянках шкіри не оброблені та не заклеєні бактерицидним пластиром.
Кожен студент працює на постійному місці та виконує завдання індивідуально. На робочому місці потрібно підтримувати зразковий порядок. Особисті речі повинні зберігатися в спеціально відведеному місці. Перед початком роботи слід одягти білий бавовняний халат та застебнути його на всі ґудзики. У халаті забороняється знаходитись за межами лабораторних приміщень, а також надівати на халат верхній одяг. Волосся необхідно прибрати з обличчя та сховати під шапочку. При роботі в лабораторії бажано знаходитися без косметики на обличчі та з коротко остриженими нігтями.
На робочому столі повинні знаходитися тільки предмети, необхідні для проведення мікробіологічних досліджень: спиртівка, пастерівські та градуйовані піпетки, пінцети, бактеріологічні петлі, шпателі, предметні та накривні скельця, пробірки, штативи, чашки Петрі, мікроскоп. На робочому місці не повинно бути сторонніх предметів (верхнього одягу, халатів, у тому числі портфелів і сумок)
Під час виконання лабораторної роботи не можна відкривати кватирки. Необхідно дотримуватися тиші, уникати зайвого руху і ходіння, відкривання і закривання дверей – всього того, що підсилює рух повітря. Виконання цих вимог запобігає прониканню сторонніх мікроорганізмів із повітря і ротової порожнини в досліджуваний матеріал.
На вході до лабораторних приміщень, де проводять біологічні дослідження встановлюють знак біологічної небезпеки та додаткову інформацію щодо відповідальних осіб. Недотримання умов ведення дослідів або порушення послідовності виконання роботи може призвести до зміни характеру кінцевих результатів, а сам хід досліджень стане неконтрольованим.
Необхідно пам’ятати, що в лабораторних роботах можуть бути використані речовини, що активно впливають на генетичний апарат клітини. Тому з цими речовинами слід поводитися обережно. Не слід допускати їх потрапляння будьяким чином на шкірні покриви.
Роботу у мікробіологічному боксі дозволено проводити лише за проходження додаткового інструктажу з техніки безпеки, наявності відповідного захисного одягу (халат, шапочка, захисна маска та захисні окуляри).
5
При роботі зі скляними предметами необхідно дотримуватись наступних вимог:
•при закриванні колби, пробірки або іншої тонкостінної посудини пробкою, тримати посудину за верхню частину шийки ближче до місця, куди повинна бути вставлена пробка, захищаючи руку рушником;
•при нагріванні пробірок над полум’ям користуватися тримачами;
•при вставленні скляних трубок у гумові пробки або шланги (при складанні приладів) попередньо змочують зовні скляну трубку і внутрішні краї шлангу або отвір у пробці водою, гліцерином або вазеліновою олією;
•при вставленні скляних трубок або термометра в просвердлену пробку, останню не впирають в долоню, а тримають за бічні сторони. Трубку або термометр тримають якнайближче до кінця, що вставляється в пробку;
•нагріту посудину не можна закривати притертою пробкою поки вона не охолоне;
•нагріваючи рідину в пробірці або інших посудинах їх тримають спеціальними утримувачами так, щоб отвір був спрямований від себе і працюючих поруч;
•при перенесенні посудин із гарячою рідиною користуються рушником, посудину при цьому тримають обома руками: однією за дно, а другою за горловину;
•великі хімічні склянки з рідиною піднімають тільки двома руками так, щоб відігнуті краї стакана спиралися на вказівні пальці;
•при закупорюванні пробками посудин із реактивами враховують їх властивості. Гумові пробки сильно набухають під дією деяких реактивів (спирт, бензол, ацетон, ефір), а під дією галогенів (бром, йод) втрачають еластичність. Такі реактиви краще закупорювати скляними притертими пробками. Луги не можна закупорювати притертою пробкою, тому що карбонати, що утворюються між пробкою і горлом, щільно заклинюють пробку;
•при переливанні рідин (крім тих, що містять біологічний матеріал) користуються лійкою;
•при змішуванні (розведенні) речовин, що супроводжуються виділенням тепла, користуються термостійким хімічним посудом;
•нагрівання сильнодіючих отруйних речовин проводять тільки в круглодонних колбах і не на відкритому вогні;
При роботі з живими культурами мікроорганізмами необхідно дотримуватись таких вимог:
•працюють з мікроорганізмами користуючись інструментом (петлею, пінцетом, ножицями тощо). Забороняється торкатися досліджуваного матеріалу руками;
•перед використанням посуд, піпетки, обладнання тощо повинні бути перевіренні на цілісність і справність;
•усі технічні маніпуляції проводять таким чином, щоб уникнути виникнення аерозолів;
6
•пробки матраців, флаконів, пробірок відкривають тільки над полум’ям пальника. Суспензію мікроорганізмів вносять в посудину так, щоб не торкатись горловини посудини. Краї отворів посудин прожарюють над полум’ям пальника і закривають пробками. Забороняється переливання рідких культур і матеріалу, що досліджується;
•при піпетуванні користуються піпетками або дозаторами. Кінець піпетки завжди повинен бути нижче рівня рідини в посудині або рідина з піпетки повинна стікати по внутрішній стінці посудини;
•обов’язкова наявність ватної пробки у тупому кінці піпетки та ватномарлевих корків у пробірках;
•центрифугування проводиться спеціально підготовленим персоналом. Якщо в процесі центрифугування розбивається пробірка, що містила суспензію мікроорганізмів, центрифугу відключають від мережі, знезаражують і очищають забруднені місця;
•всі роботи, що можуть супроводжуватися випадковими прямими контактами з мікроорганізмами виконують у гумових рукавичках;
•роботу з культурами мікроміцетів можна проводити лише за наявності індивідуальних засобів захисту дихальних шляхів;
•оскільки деякі мікроорганізми, особливо спори грибів, є алергенами, не допускається їх розпилення, тому не можна залишати відкритими чашки Петрі, пробірки, колби з культурами мікроорганізмів;
•у разі випадкового потрапляння мікробного матеріалу на шкіру або стіл чи підлогу та інші поверхні слід негайно повідомити про це викладача і в його присутності провести дезінфекцію заражених ділянок, потім обробити руки дезрозчином і ретельно вимити їх з милом;
•всі предмети, використані у роботі з живими мікроорганізмами, мають бути знезаражені фламбуванням (петлі, голки), кип’ятінням (пробірки, чашки Петрі), обробленням дезінфікуючими розчинами (шпателі, піпетки, предметні й покривні скельця).
При роботі з кислотами та лугами виконують такі заходи безпеки:
•всю роботу з концентрованими кислотами та лугами проводять під витяжною шафою, використовуючи при цьому засоби індивідуального захисту (рукавички, респіратори, гумовий фартух, захисні окуляри);
•концентровану кислоту відбирають із посудини тільки за допомогою спеціальної піпетки з грушою або сифоном;
•при приготуванні розчинів кислот, спочатку в посудину наливають необхідну кількість води, а потім помалу додають кислоту. Забороняється додавати воду в кислоту;
•при приготуванні розчинів лугів наважку лугу опускають у велику широкогорлу посудину, заливають необхідною кількістю води і старанно перемішують. Шматки лугу варто брати тільки щипцями. Щоб запобігти розігріванню розчину, при приготуванні розчинів лугів, посуд попередньо поміщають у водяну баню;
7
•розбивання великих шматків їдкого лугу на дрібні роблять користуючись захисними фартухом і рукавичками, у спеціально відведеному місці, при цьому розбиті шматки накривають бельтингом або іншим матеріалом;
•концентровані кислоти і луги виливають у раковину після попередньої їх нейтралізації;
•при кип’ятінні кислотних і лужних розчинів не можна щільно закривати посуд (пробірки і колби) пробкою до повного їх охолодження.
•при митті посуду хромовою сумішшю запобігають попаданню її на шкіру, одяг, взуття;
•при потраплянні будь-яких хімічних речовин на шкіру необхідно змити реактив великою кількістю води. Нейтралізувати вплив кислоти необхідно слабким розчином соди, а вплив лугу – слабким розчином оцтової кислоти.
При роботі з легкозаймистими речовинами (ефір, бензин, бензол, ацетон, спирт та ін.) дотримуються таких вимог:
•усі роботи проводяться у витяжній шафі при включеній вентиляції, вимкнутих газових пальниках і нагрівальних електроприладах відкритого типу;
•нагрівання легкозаймистих речовин проводять у витяжній шафі на піщаній або водяній бані з закритим електронагрівом;
•при випадковому займанні спирту необхідно терміново загасити його, закривши доступ повітря до ємкості, у якій він знаходиться;
•під час стерилізації лабораторних інструментів (шпателі, скальпелі, голки, петлі тощо) у розчині спирту необхідно слідкувати, щоб інструменти, що стерилізуються, були охолоджені.
Працюючи з відкритим полум’ям (газовий пальник, спиртівка), потрібно дотримуватися таких вимог:
•запалювати спиртівку та газовий пальник лише за допомогою сірника;
•забороняється запалювати спиртівку чи пальник запальничкою та іншою запаленою спиртівкою (газовим пальником);
•гасити запалену спиртівку потрібно, закривши доступ повітря спеціальним ковпачком, а газовий пальник – перекриттям доступу газу.
•розташовувати спиртівку потрібно на відстані не меншій ніж 20 см від краю робочого стола;
•запалену спиртівку заборонено пересувати з місця на місце;
•при випадковому займанні ватно-марлевого корку необхідно терміново загасити його, закривши доступ повітря;
•по закінченню роботи з газовими пальниками необхідно перевірити, що вихід газу перекрито.
8
Категорично забороняється:
•у лабораторію забороняється входити в головних уборах та верхньому одязі (куртка, пальто, плащ тощо), класти на столи портфелі та сумки;
•під час виконання лабораторної роботи категорично забороняється користуватися мобільними телефонами та залишати їх увімкненими;
•у лабораторії забороняється палити, вживати їжу та напої, зберігати продукти харчування;
•під час виконання лабораторної роботи студентам заборонено пересуватися по лабораторії без зайвої потреби. Всі рухи повинні бути спокійними та виваженими;
•під час роботи в лабораторії запалювати сірники, палити, включати прилади, при роботі яких може виникнути іскра;
•при роботі з мікроорганізмами заборонено торкатися обличчя, рота, носу та очей руками;
•заходити у бокс при увімкненій бактерицидній лампі;
•користуватися скляним посудом, що має сколи, тріщини, гострі краї;
•використовувати несправне електрообладнання і вмикати прилади без дозволу викладача або інженера, а також торкатися поверхні приладів мокрими руками;
•виносити за межі лабораторії будь-які матеріали і посуд, що використовується для проведення лабораторних робіт.
Після закінчення роботи необхідно:
•усі засіяні пробірки, чашки помістити у термостат або здати інженеру. Відпрацьований матеріал (пробірки, чашки Петрі, піпетки) поміщають в певні ємності за вказівкою інженера чи лаборанта для їх подальшої обробки;
•привести в порядок робоче місце: прилади ставлять на відведені для них місця, стіл протирають дезінфікуючим розчином;
•зняти халат, старанно вимити руки з милом, а за потреби обробити дезінфікуючим розчином; слід мати індивідуальний рушник або серветки для витирання рук.
9
ОРГАНІЗАЦІЯ ЗАНЯТЬ
На лабораторних заняттях з дисципліни «Генетика» студенти повинні мати халат та робочий зошит. Перед початком заняття студенти проходять інструктаж із правил роботи з обладнанням та техніки безпеки. До початку заняття студенти самостійно вивчають методику проведення експерименту та теоретичний матеріал, а також складають план проведення роботи та визначають кількість необхідного посуду, реактивів, середовищ та інших матеріалів. Викладач перед початком заняття перевіряє готовність студентів та допускає їх до виконання роботи. Студенти працюють індивідуально. Ті з них, хто виконує однакову роботу, можуть об’єднуватися у групи для підготовки культур мікроорганізмів до експерименту, посуду, середовищ, реактивів тощо.
На кожен експеримент у робочому зошиті студенти складають протокол. У ньому мають бути зафіксовані:
1.Дата, час початку та закінчення досліду.
2.Тема та мета заняття.
3.Назва об’єкта дослідження. Вік, генетичні маркери та інші мутації об’єкта досліджень, його морфологічні, фізіологічні, біохімічні та інші особливості.
4.Схема досліду (наприклад, схема схрещування, схема клонування ДНК тощо).
5.Титри суспензій мікроорганізмів, використаних у роботі.
6.Склад поживних середовищ, концентрації розчинів та буферних сумішей.
7.Температура, при якій були проведені досліди.
8.Режими роботи приладів.
9.Послідовність дій під час експерименту (при потребі час початку та закінчення його етапів) з обов’язковим наведенням схеми постановки експерименту та описом використаних методик дослідження.
10.Особливості досліду. Всі, навіть випадкові, відхилення від методики.
11.Первинний цифровий матеріал (покази приладів, результати підрахунку кількості колоній на чашці тощо) та розрахунки, зроблені на його основі; таблиці, графіки, побудовані за отриманими даними, інші ілюстрації (фотографії та їхні негативи).
12.Висновки, зроблені за результатами досліду.
Готовий протокол заняття перевіряється викладачем, проводиться захист виконаної роботи (усно або письмово) і студенту виставляється певна кількість балів згідно критеріїв оцінювання успішності навчальної роботи студентів за окремими елементами змістових модулів з дисципліни «Генетика».
10
ЛАБОРАТОРНЕ ЗАНЯТТЯ 1 ЗАГАЛЬНІ ПРАВИЛА РОБОТИ З БАКТЕРІЯМИ І БАКТЕРІОФАГАМИ
Мета: засвоєння правил пересівання та зберігання бактерій і бактеріофагів; ознайомлення та опанування методів визначення титру бактерії й бактеріофагів.
Матеріали та обладнання: термостат; чашки Петрі з повним агаризованим середовищем; пробірки з 4,5 мл фосфатного буфера; пробірки з 4,5 мл розведеного 0,5 % NaCl бульйону Хоттингера; пробірки з 3 мл 0,7 % повного агаризованого середовища; стерильні піпетки; шпателі Дригальського; суспензія культури Escherichia coli концентрацією 109 клітин/мл; суспензія бактеріофага Т4В концентрацією 109 частинок/мл.
Загальні відомості
Мікроорганізми активно використовують як об’єкти генетичних експериментів з 40-х років XX століття. Їхній вибір був зумовлений потребою вирішення важливих проблем, які з’явилися з розвитком генетики, насамперед дослідження природи гена як матеріальної структури живої клітини та його біохімічної функції, та визначив перехід до вивчення генетичних процесів на молекулярному рівні. Мікроорганізмам властиві певні особливості, що
сприяють вирішенню таких проблем:
•їх можна культивувати у строго контрольованих умовах – у поживних середовищах відомого хімічного складу, при певній температурі, аерації тощо. Це означає, що дослідник може постійно контролювати вплив умов існування організму на функціонування генів;
•мікроорганізми мають короткий життєвий цикл. Наприклад, за оптимальних умов росту час генерації (період часу між двома послідовними поділами клітини) у кишкової палички Escherichia coli становить 20–30 хв. Отже, за невеликий проміжок часу можна отримати багато поколінь нащадків певного організму. Популяції нащадків одного організму можуть становити мільйони та мільярди особин. Це важливо, бо генетичні закономірності виявляються у ряді поколінь та мають
статистичний характер. Це дає змогу виявляти рідкісні генетичні зміни, наприклад, виділяти мутанти, частота яких є дуже низькою (10-6–10-9), виявляти рекомбінанти, що виникли у результаті внутрішньо-генного кросинговеру тощо.
Убільшості мікроорганізмів одна особина – це окрема клітина. У генетиці зазвичай вивчають не окремі особини, а клонові культури мікроорганізмів (клони). Клон – це культура, що виникла у результаті нестатевого розмноження клітини. У вірусів клони – це нащадки однієї вірусної частинки. Отже, клони є групами спадково однорідних особин. Найчастіше клони отримують після посіву суспензій клітин мікроорганізмів на твердих поживних середовищах.
Окремі клітини діляться, утворюючи колонії, що і є клонами. Клони вірусів виділяють із зон лізису (негативних колоній), які виникають після посіву суспензії вірусів на газон чутливих до них культур клітин. Кожна негативна колонія утворена нащадками однієї вірусної частинки.
11
Досліджують як індивідуальні ознаки клітин мікроорганізмів, наприклад, розмір та форму клітин, особливості будови і функції їхніх органел, так і ознаки клонів. Розмір, форма, характер поверхні та забарвлення колоній, здатність культури засвоювати певні речовини як джерела живлення, чи продукувати певні речовини – це ознаки клонів. Ознаки клонів насамперед визначаються властивостями клітин, які їх утворюють. Так, наприклад, гладенькі слизисті колонії виникають завдяки здатності клітин утворювати полісахаридну капсулу. З первинного клона можна отримати вторинні клони (субклони).
Клонові ознаки зазвичай зберігаються в абсолютної більшості вторинних клонів за незмінних умов росту та розмноження. Однак у клоні можуть виникати і нагромаджуватися мутанти з новими ознаками. Тому генетична однорідність клона є відносною й тимчасовою. Клонована культура, генетична однорідність якої підтримується селекцією за певними ознаками, називається штамом.
1.1. Escherichia coli
Кишкова паличка Escherichia coli є одним з основних об’єктів генетики, молекулярної біології та генетичної інженерії. Це грамнегативна бактерія, клітини якої мають паличкоподібну форму із злегка заокругленими кінцями. Розміри: 0,4– 0,8 в ширину та 1 –3 мкм в довжину. Не утворює спор. Оптимальні умови росту: температура 30–37°С, рН 7,2–7,5. Досить добре розмножується й при кімнатній температурі. Утворює опуклі напівпрозорі колонії сіруватого кольору. Гетеротроф, факультативний анаероб, зброджує глюкозу, лактозу, мальтозу, арабінозу, галактозу, ксилозу, рамнозу та інші цукри, утворює індол та сірководень, відновлює нітрати в нітрити. Є звичайним компонентом нормальної кишкової мікрофлори людини й багатьох хребетних та безхребетних тварин. E. coli постійно виявляють й у зовнішньому середовищі – в ґрунті, воді, на різних предметах. Деякі штами E. coli є умовно-патогенними та патогенними.
Штам E. coli K-12 виділено в 1922 році в Стенфордському університеті США. Використовуючи даний штам як вихідний, було отримано більше 3000 штамів E. coli, які використовують у генетиці, молекулярній біології та біотехнології. У генетичних дослідженнях кишкову паличку вперше використали Дж. Бідл та Е. Т ейтум. Вони вивчали генетичний контроль метаболізму, використовуючи ауксотрофні мутанти E. coli. У 1946 Дж. Ледерберг та Е. Тейтум відкрили кон’югаційний процес у кишкової палички, що стало поштовхом для опрацювання методів генетичного аналізу цієї та інших бактерій. Саме у дослідах на Е. соli були встановлені основні закономірності таких важливих процесів, як реплікація, рекомбінація та репарація ДНК, мутагенез, транскрипція та трансляція, розшифровано генетичний код та механізми регулювання активності генів, опрацьовано принципи і методи генетичної інженерії.
У 1997 році секвеновано геном штаму E. coli К-12. До 2005 року секвеновано також геноми ще трьох патогенних штамів E. coli: 0157:Н7, 0157:Н7 ЕDL933 та CFT073. У клітинах E. coli є одна хромосома, яка містить кільцеву дволанцюгову молекулу ДНК. Розміри цієї молекули в різних штамів E. coli коливаються від 4,6 до 5,6 млн п.н. У штаму E. coli К-12 у хромосомній ДНК 4639221 п.н. У ній ідентифіковано 4288 відкритих рамок зчитування, що кодують
12
відомі та ймовірні білки, 7 оперонів генів рРНК (16S, 23S та 5S) та 86 генів тРНК. Гени білків становлять 87% геному, гени стабільних РНК – 0,8%, некодуючі послідовності, що повторюються – 0,7%, решта (≈11%) послідовностей виконують регуляторні та інші функції. Середній розмір гену – 951 п.н.
Для штамів Е. сoli властиві такі способи генетичного обміну, як кон’югація та трансдукція. Розроблено методики отримання компетентних клітин Е. сoli та їхньої трансформації екзогенною ДНК. Саме штами E. coli найчастіше використовують як реципієнти для клонування ДНК у генно-інженерних дослідах. Ці штами несуть низку мутацій, які роблять їх хорошими реципієнтами ДНК, а також обмежують здатність розмножуватися в кишковому тракті та виживати у природному сенредовищі поза контрольованими умовами лабораторії.
У більшості бактеріологічних лабораторій організовані музеї живих культур мікроорганізмів, що використовуються в процесі навчання, в дослідницьких, промислових та інших цілях. Правильне зберігання культур є надзвичайно важливою проблемою, якій слід приділяти таку ж увагу, як і стандартизації обладнання та вибору хімічних речовин. Втрата або зміна властивостей основних культур внаслідок їх неправильного зберігання завадили б виконанню багатьох наукових програм.
Основними цілями зберігання є підтримка життєдіяльності клітин і чистоти культур, а також попередження змін і мутацій, тобто збереження мікроорганізмів в стані, максимально близькому до початково виділеного штаму. Існує багато методів зберігання бактерій, проте не всі штами при використанні якого-небудь з них ведуть себе однаково. Вибір методу часто визначається наявністю обладнання, місця для зберігання і кваліфікованих співробітників.
1.2. Методи нетривалого зберігання мікроорганізмів
1.2.1. Субкультивування. Традиційним методом зберігання бактеріальних культур є їх періодичні пересіви на свіжі середовища. Інтервал меж пересіву залежить від мікроорганізму, використовуваного середовища і зовнішніх умов. Деякі бактерії слід пересівати через день, інші – тільки через кілька тижнів або місяців. При використанні цього методу для зберігання культур повинні бути дотримані три умови:
•відповідне підтримуюче середовище;
•ідеальна температура зберігання;
•необхідна частота пересівань.
Підтримуюче середовище. Переважно використовують мінімальні середовища, оскільки в них процеси метаболізму в мікроорганізмах йдуть із зниженими швидкостями і тому проміжки між пересівами подовжуються. Однак для росту деяких бактерій потрібні комплексні середовища, або ж для збереження їх специфічних фізіологічних властивостей необхідною є присутність в середовищі складних сполук. При використанні комплексного середовища можуть знадобитися більш часті пересівання, пов'язані з прискореним ростом бактерій або накопиченням кінцевого продукту метаболізму.
13
Зберігання. Найпростішим способом є зберігання культур мікроорганізмів при кімнатній температурі в штативі або в спеціально сконструйованому для зберігання боксі, в якому є полиці з отворами для пробірок. При такому зберіганні культур необхідний постійний контроль, оскільки вони мають тенденцію швидко висихати, і якщо умови в лабораторії не регулюються, то культури погано переносять температурні коливання. Для зменшення висихання культур використовують загвинчувальні кришки з гумовими прокладками, обгортають верхню частину пробірок парафільмом або поміщають пробірки в пластмасовий мішок. Щоб зменшити швидкість метаболізму, культури зберігають у холодильнику при 5–8 °С. Використовуючи ці запобіжні заходи, можна зберігати більшість бактерій упродовж 3–5 місяців без пересіву.
Режим пересівання. Частоту пересівань (субкультивування) визначають експериментальним шляхом. Субкультивування слід проводити якомога рідше, щоб уникнути селекції варіантів. На випадок втрати культури бажано зберігати дублікати пробірок з нею. Після кожного пересіву культуру перевіряють на чистоту і періодично проводять скорочену перевірку для виявлення будь-яких змін в фенотипових властивостях бактерій. У пересіваємих культурах не слід виділяти поодинокі колонії, оскільки при цьому підвищується вірогідність селекції мутантів.
Основними недоліками методу періодичного пересіву є: ризик зараження, помилки при позначенні штамів або наклеюванні неправильної етикетки, селекція варіантів або мутантів, можлива втрата культур і необхідність виділення спеціального місця для їх зберігання.
1.2.2. Зберігання під мінеральним маслом. Багато видів бактерій успішно зберігають місяцями або навіть роками за допомогою порівняно простого і дешевого методу, а саме під шаром стерильного медичного мінерального масла (можна використовувати вазелінове масло з питомою щільністю 0,865–0,890). Однак через недостатню стерильність мінерального масла при використанні цього методу може відбуватися зараження культур.
Масло стерилізують у сушильній шафі при 170 °С упродовж 1–2 год; автоклавування для цієї мети застосовувати не рекомендується.
Культури вирощують на скошеному агарі, в стовпчиках агару або в рідкому середовищі відповідного складу. Після появи досить гарного росту в пробірку в стерильних умовах наливають шар мінерального масла висотою не менше 2 см (культура клітин на скошеному агарі повинна бути повністю покритою) для захисту від висихання, а також для зменшення метаболічної активності і уповільнення росту культур.
Вкриті маслом культури зберігають у вертикальному положенні в холодильнику. Для перевірки збереження культур періодично визначають їх життєздатність.
Культуру під маслом можна пересівати на свіже середовище за допомогою інокуляційної голки, а отриману культуру можна знову покривати стерильним маслом. При випалюванні голки слід подбати про те, щоб бризки масла не потрапили на навколишні предмети і персонал. Первинну культуру зберігають не
14
менше кількох тижнів на той випадок, якщо субкультура виявиться забрудненою або бактерії в ній матимуть змінені властивості.
Цьому методу притаманні такі ж недоліки, як і звичайному субкультивуванню. Крім того, слід пам'ятати про можливе забруднення масла.
1.2.3. Заморожування і висушування. Зберігання бактерій в морозильній камері холодильника або звичайному морозильнику при температурі від 0° до
–20 °С призводить до різних наслідків залежно від виду бактерій. В принципі, такий спосіб зберігання не рекомендується через пошкодження клітин в евтектичних сумішах концентрованих розчинів електролітів (наприклад, в такій суміші NaCl має евтектичну точку –20 °С). Разом з тим деякі бактерії при заморожуванні можна зберігати від 6 міс до 2 років.
Більшість культур гине при висиханні в лабораторних умовах. Однак деякі культури, особливо спороутворюючих бактерій, можна зберігати роками, якщо висушувати їх у відповідному середовищі.
Ґрунт. Спороутворюючі бактерії можна успішно зберігати роками у стерильному ґрунті, висушеному на повітрі. Ґрунт стерилізують автоклавуванням по кілька годин упродовж двох днів. Інокулят з суспензії спор (по 1 мл ) вносять у пробірки, що містять стерильний ґрунт, і залишають при кімнатній температурі до помітного висихання. Потім пробірки закривають стерильними гумовими пробками і зберігають у холодильнику.
Папір. Порівняно простим і недорогим способом зберігання бактерій є їх висушування на смужках або дисках стерильного фільтрувального паперу. Цей метод ідеальний для забезпечення контролю за якістю культур. У загальній пробірці або у банці з кришкою можна зберігати багато дисків, що містять одну і ту ж культуру. При необхідності диск дістають стерильним пінцетом і в стерильних умовах вносять його у відповідне рідке середовище. Представників родини Enterobacteriaceae, а також багато інших бактерії можна зберігати на папері упродовж декількох років. Техніка цього методу полягає в наступному: стерильний папір просочують суспензією бактерій, що містить щонайменше 108 клітин/мл, і висушують на повітрі або під вакуумом. Бактерії краще виживають при висушуванні під вакуумом. Смужки або диски паперу зберігають у закритих пробірках. Якщо дані пробірки помістити в холодильник, то термін життя культури подовжується.
Желатина. Багато гетеротрофних бактерій можна зберігати у висушених краплях або дисках желатини. Зазвичай у такому вигляді бактерії краще зберігаються при температурі –20 °С, н іж при +4 °С або при кімнатній температурі. Желатинові культури готують наступним чином. Культури вирощують у відповідних середовищах і клітини отримують центрифугуванням в стерильних умовах. Осад клітин ресуспендують в малій кількості рідкого середовища, переносять у пробірку з 2-5 мл розплавленої харчової желатини і інкубують при 30 °С до досягнення щільності 108-1010 клітин/мл. Потім за допомогою стерильної пастерівської піпетки або шприца вводять краплю бактеріальної суспензії на дно стерильної пластмасової чашки Петрі. Чашку Петрі поміщають в ексикатор, що містить п’ятиоксид фосфору, і відкачують з нього повітря вакуумним насосом. Після того як краплі желатини висохнуть, їх
15
переносять з дотриманням правил асептики в стерильні пробірки з кришками і зберігають у холодильнику. Культуру бактерій отримують після перенесення желатинової краплі в стерильних умовах у пробірки, що містять відповідні середовища.
1.3. Методи тривалого зберігання мікроорганізмів
1.3.1. Ліофілізація (висушування). Висушування із замороженого стану, або ліофілізація, є одним з найбільш економічних та ефективних методів тривалого зберігання бактерій та інших мікроорганізмів. При його використанні багато фізіологічно різнорідних видів бактерій і бактеріофагів вдається зберігати в життєздатному стані 30 років і більше. Цим методом бактерії можна зберігати невеликими порціями в ампулах.
Ліофілізація полягає у видаленні води з заморожених суспензій бактерій шляхом сублімації при низькому тиску, тобто при цьому вода випаровується, минаючи рідку фазу. Висушені клітини зберігаються досить довго, якщо захистити їх від дії кисню, вологи і світла. У будь-який момент клітини можна перевести в гідратований початковий стан. Ліофілізацію проводять різними способами за допомогою різних приладів.
Обладнання. Для ліофілізації існує як просте і недороге устаткування, що включає систему ексикатора з вакуумним насосом, так і досить складне, яке є дороговартісним. Відтворюваність і збереження культур залежать від використовуваної системи. Однак часто дуже хороші результати можна отримати за допомогою простої системи, що складається з високовакуумного насосу, конденсора і камери, або розподільної гребінки.
Ампули. Існують ампули самої різної форми і розмірів; частіше інших для ліофілізації використовуються два основних типи. Для клітин, що ліофілізують в камері, рекомендуються подвійні ампули. Такими ампулами легше користуватися, при їх використанні зменшується ймовірність інфікування сторонніми мікроорганізмами і такі ампули є більш надійні для зберігання патогенних штамів, ніж звичайні одинарні ампули, які сушать і закривають безпосередньо перед вставлянням у гребінку.
Підготовка культури. Успіх ліофілізації залежить від якості використовуваних клітин, від того, наскільки вони життєздатні і в яких умовах вони виросли. Вирощують культуру клітин так, щоб в суспензії містилося не менше 108 клітин/мл. Клітини збирають у період максимальної стабільності і життєздатності культури, тобто в пізній експоненційній або ранній стаціонарній фазах росту.
Кріопротектори. Для підготовки клітин до ліофілізації їх суспендують в середовищі, що містить кріопротектор. У якості кріопротекторів використовують 24 % розчин сахарози, розведений рівним об'ємом поживного середовища (кінцева концентрація сахарози 12 % при використанні одинарних ампул) або декстран (10% ), кінську сироватку, інозит та ін.
У разі використання подвійних ампул застосовують наступну процедуру. Готують 20 % зняте молоко і стерилізують його порціями (по 5 мл) упродовж 20 хв при 116 ° С. У стерильних умовах збирають клітини, які виросли на поверхні
16
агару, змиваючи їх 20 %-ним знятим молоком. Клітини, вирощені в рідкій культурі, відокремлюють в стерильних умовах і потім суспендують осад у стерильному молоці, так щоб вийшла суспензія, що містить щонайменше 106 кл/мл. Цю ж процедуру застосовують і в тому випадку, коли в якості кріопротектору використовується сахароза.
У кожну подвійну ампулу наливають 0,2 мл суспензії клітин, закривають їх ватними пробками і підрівнюють пробки ножицями. Після приготування суспензії її слід розливати по ампулам якомога швидше. Інтервал між розливом і процесом ліофілізації має бути зведений до мінімуму, щоб уникнути змін у культурі.
При використанні одинарних ампул в кожну з них наливають 0,1 мл суспензії бактерій. Вставляють ватні пробки приблизно на 1,3 см нижче краю ампули і обпалюють їх верхню частину, щоб прибрати зайві волокна вати.
Зберігання. Культури, ліофілізовані як у подвійних, так і в одинарних ампулах, зберігають при температурі нижче +5 °С. При кімнатній температурі ліофілізовані культури зберігати не можна .
Відновлення культур з ліофілізованих клітин. Сильно нагрівають запаяний кінець зовнішньої ампули на пальнику і швидко капають на нього 1-2 краплі води, щоб гаряче скло тріснуло. Обережно, але швидко пінцетом надламують і видаляють верхівку, а потім витягують пробку зі скловолокна і виймають внутрішню ампулу. З останньої акуратно видаляють ватну пробку, намагаючись не допустити забруднення культури зовнішніми волокнами.
Перш ніж відкрити одинарні ампули, їх надрізають тригранною пилочкою на відстані близько 2,5 см від верхнього кінця, а потім дезінфікують, протираючи шматочком марлі, просоченої 70 %-ним спиртом. Обгортають ампулу стерильною марлею і обламують надпиленний кінець. Зазвичай це роблять у витяжній шафі, а
у разі патогенних бактерій – у спеціальному закритому боксі. Перед тим як внести
вампулу рідке середовище, обламаний кінець злегка обпалюють. Деякі дослідники використовують вольфрамовую голку, нагріту в киснево-газовому пальнику. Такою голкою їм вдається зробити у верхній частині ампули маленький отвір. Це забезпечує більш повільне проникнення повітря в ампулу і стерильність простору навколо отвору. Рідке середовище вносять в ампулу стерильним шприцом з голкою і витягують суспензію бактерій ним же.
Ліофілізовану культуру переводять в суспензію відразу після відкриття ампул, додаючи в кожну 0,3 – 0,4 мл відповідного стерильного рідкого середовища. Суспензію в ампулах добре перемішують і переносять у пробірки з 5 мл рідкого середовища. Після ретельного перемішування відбирають 0,2 мл суспензії та наносять на щільне або напіврідке середовище того ж складу. Необхідно перевіряти чистоту культури до ліофілізації і після неї. Для цього суспензію клітин стерильно розводять і роблять посів штрихом на щільні середовища. Пробірки та чашки з середовищем інкубують при оптимальній для бактерій температурі і, як тільки починається їх ріст, роблять пересів на свіже середовище, щоб переконатися в чистоті культури. Ріст бактерій, що піддавалися ліофілізації, часто починається після тривалої лаг-фази. Тому не можна робити висновок про загибель культури , якщо інкубація була недостатньо тривалою.
17
Перевірка життєздатності бактерій. Щоб визначити, наскільки ефективний процес висушування бактерій в замороженому стані, перевіряють їх життєздатність як до, так і після ліофілізації. Крім того, слід проводити періодичні перевірки, що підтверджують життєздатність зберігаємих ліофілізованих культур. Їх бажано також перевіряти на випадок появи будь-яких змін у властивостях бактерій в результаті їх заморожування – висушування або тривалого зберігання.
1.3.2. Ультразаморожування. Деякі види бактерій, що не піддаються ліофілізації, вдається довгостроково зберігати в замороженому стані при температурі рідкого азоту (–196 °С) або в його парах (–150 °С) в добре ізольованих резервуарах. Таким способом можна успішно зберігати безліч «вибагливих» бактерій, які не втрачають при цьому своїх фенотипових властивостей упродовж 15 років.
Бактеріальні культури можна також досить тривало зберігати в морозильниках з дуже низькими температурами (при –70 °С). Такий метод зберігання цілком придатний для багатьох бактерій. Однак при цьому необхідно пам'ятати, що іноді вимикається електроживлення мережі або виходить з ладу компресор. У таких випадках втрату цінних колекцій допомагають запобігти сигнали попередження, а також запасні морозильники або електричні генератори. Наявні в даний час у продажу морозильники зазвичай обладнані аварійною сигнальною системою, що працює на акумуляторі.
Рекомендується також використання додаткових систем попередження, таких, як звуковий, чутливий до відключення напруги датчик температури, що працює в інтервалі від – 75°С до 200°С ( ± 0,3°С). Він працює від акумулятора і може бути закріплений на стіні.
Зберігання в рідкому азоті. Вважається, що використання рідкого азоту для тривалого зберігання мікроорганізмів обходиться дуже дорого. Однак дані останніх років про успішне зберігання в рідкому азоті фізіологічно різнорідних бактерій свідчать про те, що його використання є рентабельним, незважаючи на високу вартість.
Нині випускається ряд холодильників, що працюють на рідкому азоті, різної конструкції і ємності, наприклад типу MVE, Cryogenics, Linde, Union Carbide Corp. Їх місткість варіює від 10 до 1000 л і в них можна зберігати від 300 до 40 000 ампул з культурами клітин.
Ампули. Випускаються різні види ампул для зберігання бактерій в рідкому азоті. Особливо широко використовуються такі типи: боросилікатні з товстими стінками та спеціальні широкогорлі ампули. Ці ампули ємністю 1,2 мл мають вдавлення, що полегшують їх відкривання. Замість скляних ампул можна використовувати попередньо простерилізовані поліпропіленові пробірки з загвинчуємими кришками і силіконовими прокладками.
Кріопротектори. При зберіганні бактерій в рідкому азоті застосовують кріопротектори двох типів. До першого належать гліцерин і диметилсульфоксид (ДМСО), які легко проходять через клітинну мембрану і забезпечують як внутрішньоклітинний, так і позаклітинний захист від заморожування. До другого виду кріопротекторів відносяться такі речовини, як сахароза, лактоза, глюкоза,
18
маніт, сорбіт, декстран, полівінілпіролідон і полігліколь, які забезпечують захисну дію на зовнішній поверхні клітинної мембрани. Протектори першого типу, тобто гліцерин і ДМСО, виявилися більш ефективними і придатними для широкого кола бактерій. Вибір кріопротектора залежить від виду бактерій. При заморожуванні нових штамів слід попередньо перевірити дію на них кріопротектора.
Гліцерин і ДМСО зазвичай додають у середовище вирощування у об’ємній частці 10 і 5 % відповідно. Гліцерин стерилізують автоклавуванням 15 хв при 104 Па.
Стерилізацію ДМСО здійснюють фільтруванням, використовуючи пористі свічки Села з розміром пор 03. ДМСО збирають порціями по 10-15 мл в стерильні пробірки і зберігають у замороженому стані при 5°С (ДМСО замерзає при 18 °С). Через накопичення продуктів окисного розпаду ДМСО у відкритих бутлях зберігають не більше 1 місяця.
Підготовка культури. У виживанні бактерій після заморожування рідким азотом важливу роль відіграє фізіологічний стан культури. В основному в замороженому стані зберігають культури клітини, зібрані у середині або в кінці експоненційної фази росту.
Клітини вирощують у відповідному середовищі. У разі рідких культур клітини відокремлюють у стерильних умовах центрифугуванням і ресуспендують осад у стерильному свіжому середовищі, що містить або 10 % гліцерину (готують додаванням 20 %-ного гліцерину до рівного об'єму стерильного середовища), або 5% за об’ємом ДМСО (приготованого додаванням відповідної кількості 100% ДМСО до стерильного середовища). У разі вирощування культур клітин на агаризованому середовищі, клітини змивають з його поверхні стерильним рідким середовищем, що містить відповідний кріопротектор, і розливають у стерильні марковані ампули по 0,4 мл суспензії, що містить не менше 108 клітин/мл.
Якщо штам є патогенним, ватяну пробку зрізають на рівні верхнього кінця ампули і зберігають ампули в парах рідкого азоту незапаяними. Це роблять, щоб уникнути нещасних випадків у процесі їх запаювання або щоб уникнути розриву ампули в процесі відтаювання (через потрапляння в ампулу рідкого азоту через дрібні отвори і подальшого швидкого виходу газу при кімнатній температурі).
У разі непатогенних бактерій ампулу охолоджують при 4°С не менше 30 хв. Потім нагрівають над місцем кругової насічки (надрізом) скла, обертаючи в полум'ї упродовж декількох секун , щоб видалити вологу. Ця процедура дозволяє уникнути збільшення тиску всередині ампули, яке може призвести до появи бульбашок в процесі її запаювання. Перед заповненням з ампули знімають ватну пробку. Бажано запаювати ампули за допомогою обладнаного киснево-газовим пальником напівавтоматичного пристрою, призначеного для витягування й запаювання скла. Вважається, що в цьому випадку гарантується мінімум дрібних капілярних пор. Обережно пінцетом гарячу ампулу ставлять у штатив, занурений в баню з холодною водою, з таким розрахунком, щоб вона була занурена на глибину 0,6 см. Не можна допускати попадання води на гарячу частину ампули, інакше скло може тріснути. Після охолодження ампули готові до заморожування. Якщо всі операції були виконані задовільно, температура вмісту в ампулах не повинна підніматися вище 25°С. За відсутності автоматичного пристрою ампули
19
запаюють вручну на газовому пальнику, але їх необхідно при цьому перевіряти на герметичність. З цією метою ампули після запаювання залишають на 30 хв при 5°С в 0,05 %-ному розчині метиленового синього. Погано запаяні ампули виявляють за проникненням в них фарби.
Заморожування. На сьогодні опубліковано багато робіт про вплив швидкості охолодження в процесі заморожування на виживання бактерій. Найкращі результати з виживання та відновлення бактеріальних культур були отримані в разі повільного охолодження клітин, наприклад, зі швидкістю 1°С/хв .
Відтаювання. При витягуванні заморожених скляних ампул з рідкого азоту не виключена можливість їх вибуху, тому під час цієї процедури слід надягати рукавички і захищати екраном обличчя. Щоб оживити заморожені культури, їх швидко розморожують у водяній бані при 37°С і слабкому струшуванні, поки не розтане весь лід. У разі скляних ампул для цього зазвичай потрібно 40-60 с, а в разі поліпропіленових – 60-120 с. Відразу ж після відтаювання ампулу витягують з водяної бані і дезинфікують її 70 %-ним етанолом. Ампулу розкривають і в стерильних умовах переносять культуру в свіже середовище. Для визначення ефективності методу зберігання в разі кожного штаму перевіряють життєздатність культури. Передбачуваний час виживання різних бактерій наведено у табл. 1.1.
1.4. Типи поживних середовищ для вирощування мікроорганізмів
Поживне середовище для культивування мікроорганізмів повинно містити всі елементи, з яких будується клітина, причому в доступній для засвоєння організмом формі. Такими основними компонентами поживного середовища повинні бути: джерело вуглецю та енергії, джерела мінеральних сполук (азот, фосфор, сірка, калій, магній, кальцій, залізо та мікроелементи), а також (у разі потреби) і фактори росту.
За складом середовища поділяються на дві групи: синтетичні та натуральні (складні). У багатьох особливо вимогливих мікроорганізмів потреби в поживних речовинах поки що досліджені недостатньо. Тому їх вирощують на середовищах, що містять пивне сусло, морквяний та сливовий соки, м’ясний екстракт, сінний відвар, кокосове молоко, дріжджовий екстракт. Для зниження вартості до поживних розчинів замість чистих сполук додають складні суміші, такі як молочна сироватка, патока, меляса, кукурудзяний екстракт та ін. Такого роду середовища називають складними. Середовища, які складаються з продуктів рослинного та тваринного походження, називають також натуральними. До натуральних середовищ невизначеного складу належать і напівсинтетичні середовища, до складу яких входять сполуки відомої хімічної природи і речовини невизначеного складу. До напівсинтетичних середовищ належать м’ясопептонний бульйон з глюкозою та фосфорнокислим калієм, картопляне середовище з глюкозою та пептоном.
За призначенням розрізняють елективні та диференційно-діагностичні
середовища. Елективні забезпечують переважний розвиток одного виду або групи мікроорганізмів і менш придатні (зовсім непридатні) для розвитку інших. Диференційно-діагностичні (індикаторні) дають змогу досить швидко відрізнити одні види мікроорганізмів від інших.

20
Таблиця 1.1
Час виживання бактерій при використанні різних методів їх зберігання
|
Частота |
|
Час виживання, роки |
|
||
|
Під |
|
При |
|
|
|
Рід |
* |
|
|
|
||
пересівів , |
мінераль- |
У стериль- |
глибокій |
Після |
У рідкому |
|
|
місяці |
ним маслом |
ному грунті |
заморозці |
ліофілізації |
азоті |
Acetobacter |
|
1 |
|
1–3 |
>30 |
>30 |
Achromobacter |
1 |
1–2 |
|
1–3 |
>30 |
>30 |
Acinetobacter |
0,25 |
|
|
|
>30 |
>30 |
Actinobacillus |
0,25 |
2–3 |
|
|
>30 |
>30 |
Actinomyces |
1. |
|
1–2 |
2–3 |
>30 |
>30 |
Agrobacterium |
1–2 |
1–2 |
1–2 |
|
>30 |
>30 |
Arthrobacter |
1–2 |
|
|
1–2 |
>30 |
>30 |
Bacillus |
2–12 |
1 |
1–2 |
2–3 |
>30 |
>30 |
Bacteroides |
0,25 |
|
|
1 |
>30 |
>30 |
Bifidobacterium |
0,25 |
|
|
|
>30 |
>30 |
Chromatium |
1 |
|
|
|
>6 |
>10 |
Clostridium |
6–12 |
1–2 |
|
2–3 |
>30 |
>30 |
Corynebacterium |
1–2 |
1 |
|
1–2 |
>30 |
>30 |
Enterobacterium |
1–4 |
1–2 |
|
|
>30 |
>30 |
Escherichia |
1–4 |
1–2 |
|
|
>30 |
>30 |
Erwinia |
1–4 |
1–2 |
|
|
>30 |
>30 |
Flavobacterium |
1 |
2 |
|
|
>30 |
>30 |
Gluconobacter |
1 |
|
|
|
>30 |
>30 |
Haemophilus |
0,25 |
0,08 |
|
|
>30 |
>30 |
Klebsiella |
1–4 |
1 |
|
1–2 |
>30 |
>30 |
Lactobacillus |
0,25 |
|
|
|
>30 |
>30 |
Methanobacterium |
1 |
|
|
|
>10 |
>10 |
Methanomonas |
1 |
|
|
|
>10 |
>10 |
Micromonospora |
1 |
|
1–2 |
1 |
>30 |
>30 |
Neisseria |
1 |
|
|
|
>30 |
>30 |
(N. gonorrhoeae) |
0,25 |
0,08 |
|
|
>30 |
>30 |
(N. meningitidis) |
0,25 |
0,08 |
|
|
>30 |
>30 |
Nocardia |
1–4 |
1 |
|
1–2 |
>30 |
>30 |
Proteus |
1–2 |
1 |
|
1–2 |
>30 |
>30 |
Pseudomonas |
1–3 |
|
|
|
>30 |
>30 |
Spirillum |
0,25 |
0,5 |
|
1 |
>30 |
>30 |
Staphylococcus |
1–2 |
|
|
1 |
>30 |
>30 |
Streptococcus |
1–2 |
1 |
|
|
>30 |
>30 |
Streptomyces |
1–8 |
1–2 |
2–3 |
1–3 |
>30 |
>30 |
*Частота пересівання залежить від використаного середовища зберігання
21
За фізичним станом розрізняють рідкі, щільні та сипкі середовища.
Прикладами сипких середовищ є розварене пшоно, висівки, кварцевий пісок, насичені поживним розчином. Для виготовлення щільних середовищ, на яких мікроорганізми ростуть у вигляді колоній, до рідких поживних розчинів додають речовини, які надають їм гелеподібної консистенції. Желатину використовують дуже рідко, оскільки вона має низьку температуру плавлення – (26–30 °С) і, крім того, розкладається багатьма мікроорганізмами. Майже ідеальним засобом для одержання щільних середовищ є агар. Агар – це полісахарид, який виділяють з червоних морських водоростей. Його розчини плавляться при 100 °С, а при 44 °С перетворюються на твердий прозорий гель. Розкладати агар здатні тільки деякі бактерії. Концентрація агару у середовищі становить 15–20 г/л. Якщо потрібні щільні середовища, які не містять органічних речовин, для затвердіння використовують силікагель.
1.5. Елективні методи культивування (накопичувальні та чисті культури)
Багато які мікроорганізми дуже легко виділити з навколишнього середовища, для них без особливих ускладнень можна підібрати умови, які б забезпечували їх ріст. Проте існує дуже багато мікроорганізмів, про які стало відомо лише після того, як була розроблена техніка накопичувальних культур. Честь цього відкриття належить нашому співвітчизнику С.М. Виноградському та голландському вченому М.В. Бейєрінку.
Накопичувальні культури. Метод накопичувальних культур дуже простий. Для накопичення потрібні такі умови, в яких організм долає конкуренцію інших. Підбираючи ряд факторів (джерела енергії, вуглецю, азоту, акцептори електронів, газову атмосферу, освітленість, температуру, рН та ін.), створюють певні умови та інокулюють (засівають) середовище змішаною популяцією, яка, наприклад, є в ґрунті, мулі, воді тощо. Найпристосованіший до цих конкретних умов мікроорганізм росте і витісняє всі інші супутні організми. Багаторазовими пересівами на такому самому рідкому поживному середовищі і посівом на агаризоване середовище такого самого складу можна без особливих зусиль виділити накопичений штам. Найкращим матеріалом для інокуляції є проби з тих місць, де вже є «природне збагачення». Наприклад, якщо потрібно виділити мікроорганізми, які ростуть на вуглеводнях, найкраще відбирати пробу з ґрунту на нафтопромислах або з нафтових відстійників. Екстремально термофільні бактерії з великою ймовірністю можна виділити з води гарячих джерел, анаеробні – з мулових відкладень.
Метод накопичувальних культур дає можливість виділяти мікроорганізми з будь-якою комбінацією потреб в поживних речовинах, якщо такі організми взагалі існують у природі. Наприклад, мінімальне середовище, яке не містить азоту, при освітленні є вибірковим для азотфіксувальних ціанобактерій. Якщо це саме середовище доповнити органічним джерелом енергії та вуглецю, то в темноті в аеробних умовах буде розвиватись азотобактер, а в анаеробних – клостридії. Дуже часто разом з «позитивною» селекцією здійснюється і «негативна». Наприклад, на середовищі, яке містить азид натрію (дихальна
22
отрута), у присутності кисню виростуть молочнокислі бактерії (факультативні анаероби), а ріст аеробів пригнічується. Для пригнічення росту грампозитивних бактерій у середовище додають пеніцилін. Ріст мікроміцетів пригнічується в присутності антибіотика ністатину.
Чиста культура. Це потомство однієї-єдиної клітини (клон). Чисті культури мікроорганізмів виділяють в основному на агаризованому середовищі (на його поверхні чи всередині). Найпростіший спосіб одержання чистих культур аеробів – посів зразків з накопичувальної культури на поверхню щільного середовища методом виснаженого штриха. Чисті культури аеробів можна отримати також за методом Коха (метод посіву на поверхню щільного середовища послідовних десятикратних розведень клітинної суспензії), а анаеробів – методом посіву послідовних десятикратних розведень клітинної суспензії в розплавлений агар (температура 45 °С) і вирощування без доступу повітря.
1.6. Ріст бактерій у бактеріальній популяції
Популяція – це сукупність бактерій одного виду (чиста культура) або різних видів (змішані культури, асоціації), що розвиваються в обмеженому просторі (поживне середовище та ін.).
У бактеріальній популяції постійно відбувається ріст бактерій, розмноження та відмирання клітин. Для спостережень за розвитком бактеріальної популяції визначають:
•концентрацію бактерій – кількість клітин в 1 мл клітинної суспензії (культуральної рідини);
•бактеріальну масу (маса клітин, біомаса, густина бактеріальної маси) – кількість міліграмів сухої біомаси в 1 мл клітинної суспензії (культуральної рідини).
Під час росту періодичної (статичної) бактеріальної культури (тобто такої культури, яка вирощується в будь-якому середовищі без його змін) не може бути кореляції між цими двома показниками. Ці показники необхідно розрізняти.
У популяції не всі клітини життєздатні. Живими вважаються ті клітини, які можуть утворювати колонії на (в) агаризованому середовищі або суспензію в рідкому поживному середовищі. Ці життєздатні клітини виявляють спеціальними методами, призначеними для визначення кількості живих клітин. У загальну кількість клітин входять живі, пошкоджені та мертві клітини. Тому розрізняють
методи визначення загальної кількості клітин і кількості живих клітин.
Загальна кількість клітин. Існує кілька методів визначення загальної кількості клітин:
•найпоширенішим є підрахунок загальної кількості клітин під мікроскопом у тонкому шарі за допомогою лічильної камери;
•використання стандартів мутності. Це один із найдавніших методів, суть якого полягає у порівнянні мутності досліджуваної клітинної суспензії з мутністю стандартних розчинів, для яких відома концентрація клітин;
23
•використання електронного лічильника. Його дія ґрунтується на зниженні провідності розчину електролітів під час проходження однієї бактерії через вузький отвір;
•якщо кількість клітин становить менше як 106 на 1 мл, використовують
метод мембранних фільтрів (зразок фільтрують через мембранний фільтр, потім цей фільтр сушать, фарбують, просвітлюють і підраховують кількість клітин під мікроскопом).
Кількість живих клітин. Визначається за методом Коха (посів послідовних десятикратних розведень клітинної суспензії на чашки Петрі з агаризованим середовищем). Кількість життєздатних клітин визначають за кількістю колоній, які виросли на чашках (іноді користуються терміном колонієутворююча одиниця, або КУО).
Біомасу визначають за допомогою прямих і непрямих методів. У повсякденній практиці перевага віддається непрямим методам (з використанням відповідного калібрувального графіка).
Прямі методи. Існує кілька прямих методів визначення біомаси:
•сиру біомасу визначають після осадження клітин центрифугуванням. Після висушування відмитих клітин можна визначити суху біомасу;
•визначення загального азоту (наприклад, за методом К’єльдаля) або загального вмісту вуглецю (наприклад, за методом Тюріна);
•визначення вмісту білка (наприклад, за допомогою біуретового методу,
методу Лоурі чи Фоліна).
Непрямі методи. Відомо кілька таких методів:
•біомасу визначають за оптичною густиною клітинної суспензії з наступним перерахунком на суху біомасу за допомогою калібрувального графіка;
•визначення показників інтенсивності метаболізму, безпосередньо пов’язаних з ростом (поглинання кисню, утворення СО2).
1.7. Пересівання й зберігання бактерій і бактеріофагів
У генетичних дослідженнях мікроорганізмів велике значення мають якісні показники висіваємих культур бактерій і бактеріофагів. Техніка висіву бактерій багато в чому подібна до загальноприйнятих правил. Однак є ряд істотних відмінностей:
•одержання генетично чистої культури, тобто очищеної від супутніх мутантів і ревертантів;
•відбір монотипових колоній за морфологічними і фізіолого-культурними ознаками після висіву бактерій на спеціальні середовища;
•дотримання фізичних умов вирощування бактерій (температура, період культивування, доступ кисню, щільність середовища, освітленість) .
Пересівання музейних культур варто робити якомога рідше (не частіше двох разів на рік), тому що під час пересівань завжди існує небезпека потрапляння мутантів або ревертантів, що виникають у культурі, у пересіваний матеріал. У ході послідовних пересівів вони можуть поступово витісняти первісну культуру.
24
У процесі ведення музейних культур варто періодично очищати їх від можливих мутантів або ревертантів відбором матеріалу із клонів, тобто окремих колоній, що виросли з одиничних клітин, або спор одночасно з перевіркою їхніх генотипів.
Бактеріофаг вирощують на генетично чистих культурах зараженням їх на рідкому або твердому поживному середовищі із наступним відмиванням фага від субстрату й фільтрацією через мембранні мікропористі фільтри. Фаги містяться в рідкій фазі. Зберігання генетично чистих клонів бактерій і бактеріофагів – відповідальний етап генетичних досліджень. Тому і до їх вирощування, і до їх зберігання ставляться жорсткі вимоги.
Метод зберігання бактеріофага залежить від його виду. Так, бактеріофаг Т4 зберігають у стерильному бульйоні в герметично закритих пробірках або флаконах при температурі 0 – 4 °С до кількох років. Бактеріофаг P1 кс зберігають у стерильному буфері з хлористим кальцієм у холодильнику.
Фаг досить швидко втрачає активність, тому для його зберігання використовують суспензії з високим титром: 1010 – 1011 частинок/мл. Пересівають фаг P1 кс один – два рази на рік.
Багато видів бактеріофагів можна зберігати й за звичайних температурних умов у водному середовищі або середовищі фізіологічного розчину без видимих порушень генетичних ознак протягом досить тривалого періоду часу.
Поживні середовища, використовувані в генетичних дослідженнях на клітинах мікроорганізмів, в основному не відрізняються від звичайних середовищ, однак слід зважати на те, що:
•хімічні речовини, використовувані для приготування середовищ, мають бути високого ступеня чистоти;
•поживні середовища слід готувати тільки на дистильованій воді і з компонентів, які були попередньо роздільно простерилізовані;
•у деяких випадках для стерилізації середовищ застосовують методи фільтрації й виморожування;
•для приготування середовищ потрібно чітко дотримуватись дозування компонентів, температури й тривалості стерилізації;
•всі застосовувані середовища – синтетичні;
•багато поживних середовищ є дефіцитними по окремих компонентах живлення, тобто є диференційованими;
•досить часто поживні середовища використовують у вигляді двоабо багатошарового субстрату; при цьому як за складом, так і за консистенцією вони можуть бути неоднакові;
•у практиці генетичних досліджень застосовують правило підсушування поживних середовищ (видалення конденсаційної вологи), яке, в основному, стосується середовищ, розлитих у чашки Петрі; підсушування сприяє швидкому усмоктуванню мікробної суспензії; його проводять в сушильній шафі при температурі не більше 60 °С звичайно протягом 1 год.
1.8.Визначення титру бактерій і бактеріофагів. Визначення титру бактерій і бактеріофагів у генетичних дослідженнях необхідно для встановлення

25
генетичної взаємодії між бактеріями й фагом, активності репродукції фага, ступеня стійкості бактерій до дії фага тощо.
Для визначення титру бактерій використовують звичайні мікробіологічні методи (висів розведень на чашки Петрі). Для визначення титру бактеріофага розроблено ряд методик одержання фагових лізатів. Одна з них ґрунтується на ефекті утворення зон просвітління (бляшок), які відповідають кількості інфекційних фагових частинок, що містяться в лізаті.
В основі методу розведень лежить титрування суспензії мікроорганізмів і наступний розсів її на тверде поживне середовище. Вважають, що кожна життєздатна клітина, спора або вірусна частинка утворить окрему колонію. Підрахувавши число вирослих на чашках колоній і знаючи розведення й об'єм висіяних проб, можна обчислити кількість життєздатних одиниць в 1 мл вихідної культури (суспензії) за формулою:
N = Vdn ,
де n – середня кількість колоній на чашці (для даного розведення); d – розведення; V – об'єм висіваної проби, мл.
Зручне для підрахунку число колоній на звичайно використовуваних чашках Петрі (діаметр 90 – 100 мм) становить: для бактерій – 50...200; цвілевих грибів – 20...50; бактеріофагів – 200...600.
Успіх аналізу забезпечують:
•точність у відборі проб і розведень;
•разове використання піпеток;
•гомогенність суспензії культури;
•підсушування середовища при висіві рухливих форм мікроорганізмів (наприклад E. соli);
•суворий контроль температури й складу розріджувача (вода, дистилят, буфер);
•запобігання інактивації бактеріофагів і вірусів у сольових розчинах, які не містять білку або желатини при їхньому розведенні або струшуванні.
Для визначення титру використовують два методи – поверхневий і глибинний (двошаровий). За першого – на агарову пластину вносять піпеткою 0,1 мл суспензії, яку ретельно розтирають шпателем. За другого – суспензію з розведенням піпеткою вносять у розплавлений і охолоджений до температури 45
– 48 °С напіврідкий 0,7 % агар і після перемішування виливають на поверхню більш щільного (2 % агар), попередньо підсушеного, середовища в чашці Петрі. Звичайно в 3 – 5 мл напіврідкого агару вносять 0,5 мл розведеної суспензії.
Бактеріальні клітини розводять у 0,05 М фосфатному буфері з рН – 7,0. Як повне агаризоване середовище застосовують агаризований бульйон Хоттингера. У разі висіву клітин двошаровим способом використовують напіврідке 0,7 % і щільний 2 %, при поверхневому способі – 2 % повне агаризоване середовище.
Фагову суспензію розводять вдвоє у розбавленому вдвоє бульйоні Хоттингера з 0,5 % NaCl. Штамом-хазяїном є культура E. соli. Для висіву проб з розведень використовують 0,7 % ПА і 2 % повне агаризоване середовище.
26
Завдання на виконання
І. Розведення й розсів бактеріальної культури.
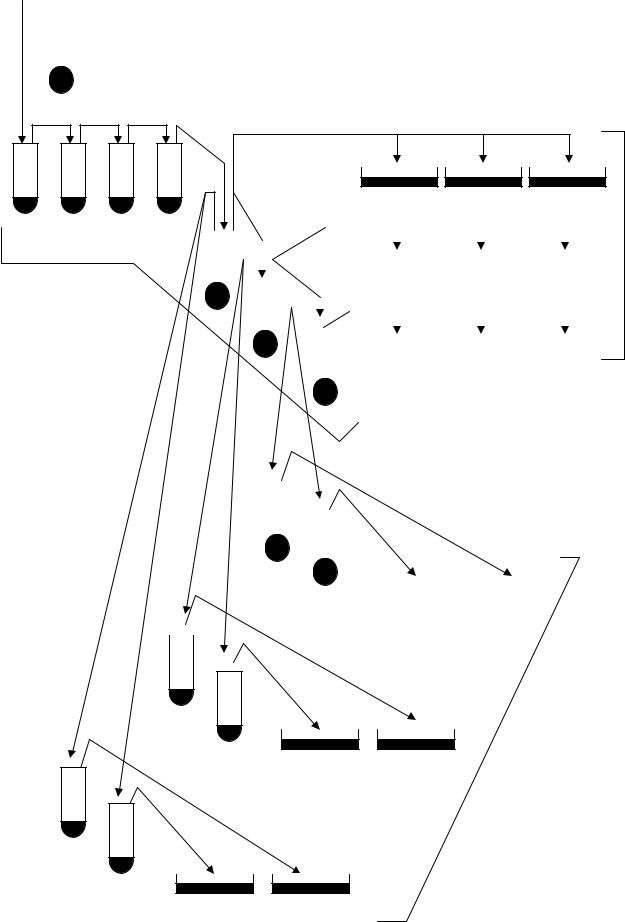
1. На занятті використовують нічну культуру, в ирощену на поживному середовищі концентрацією 109 клітин/мл. Із цієї культури готують послідовні розведення (до семи розведень), для чого в кожну пробірку вносять по 4,5 мл фосфатного буфера (рис. 1.1). З вихідної культури відбирають по 0,5 мл (перед відбором проб суспензія ретельно перемішується) і переносять у першу пробірку, не занурюючи піпетку в буфер. Культури розводять за загальноприйнятою схемою. Розсів бактеріальної культури проводять двома способами – поверхневим
ідвошаровим.
2.При поверхневому розсіві з п'ятого, шостого та сьомого розведень беруть проби по 0,1 мл і обережно, не ушкоджуючи агар, наносять на чашки з повним агаризованим середовищем. Суспензію рівномірно розтирають по всій поверхні середовища мікробіологічним шпателем. Якщо аналіз виконують на паралельних чашках, з кожного розведення розтирання суспензії проводять тим самим шпателем.
3.При двошаровому методі розсіву з пробірок 5, 6 і 7 відбирають проби по 1 мл і вносять по 0,5 мл у дві пробірки, що містять по 3 мл розплавленого напіврідкого агару (пробірки з агаром мають перебувати у водяній бані при температурі 46 °С). Суспензію в агарі швидко перемішують струшуванням і виливають на чашки із твердим поживним попередньо підсушеним середовищем. Розлите середовище треба рівномірно розподілити по поверхні агару. Процедура розсіву триває 5 – 7 с. Після застигання агару (10 – 15 хв) чашки в переверненому стані поміщають у термостат з температурою 37 °С й інкубують протягом 24 год.
ІІ. Розведення й розсів суспензії бактеріофага.
1.Використовується вихідна суспензію бактеріофага Т4В концентрацією 109 частинок/мл, приготовлену розведенням фаголізату. Розведення бактеріофага
проводять у ряді з семи пробірок, які містять по 4,5 мл розведеного 0,5 % NaCl бульйону (рис. 1.2). Вихідну суспензію бактеріофагу розводять до 10–7 частинок/мл.
2.Висів бактеріофага проводять двошаровим методом. Для цього з розведення 10–6 і 10–7 відбирають проби по 1 мл і вносять по 0,5 мл у кожну з двох пробірок з 3 мл розплавленого напіврідкого агару (при температурі 46 °С).
Напіврідкий агар попередньо заражають бактеріями, для чого в кожну пробірку вносять по 0,1 мл суспензії E. соli з титром 109 клітин/мл. Вміст кожної пробірки перемішують і виливають у чашку – верхній шар – на нижній шар підсушеного 1,2 % повного агаризованого середовища. Після застигання верхнього шару чашки поміщають у переверненому вигляді в термостат і витримують протягом 18 – 24 год при температурі 28 °С.
|
|
|
0,5 |
Вихідна бактеріальна культура |
27 |
|
|
|
|
|
|
з титром |
|
|
|
|
|
|
|
|
|
|
|
|
|
109 кл/мл |
|
|
|
|
|
|
|
Поверхневий розсів |
|
|
|
|
|
|
|
0,5 |
0,5 |
0,5 |
По 0,1 мл на чашку |
|||
|
|
|
|
|
0,5 |
|
0,5 По 0,1 мл на чашку
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
по 4,5 мл буфера |
|
|
|
|
|
|
|
|
0,5 |
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
|
|
|
|
По 0,1 мл на чашку |
|
|
|
||||||||||||||||||
|
|
|
|
||||||||||||||||||||||
|
|
|
|
|
|
||||||||||||||||||||
рН 7,0 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
0,1
0,1 |
Двошаровий розсів |
0,1 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
||
|
3 мл |
|
|
|
|||||
|
|
|
|
||||||
|
|
|
|||||||
|
|
0,7 % ПА |
|
|
|
||||
|
|
0,1 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
0,1
0,1
3 мл |
2 % ПА |
0,7 % ПА |
3 мл
0,7 % ПА
Рис.1.1. Схема титрування й розсіву суспензії E. соli (ПА – повне агаризоване середовище)
2 % ПА

0,5 мл
Вихідна суспензія бактеріофага
Т4В з титром 109 частинок/мл
0,5 0,5 0,5 0,5
0,5 0,5
0,5
0,5
0,5
По 4,5 мл розведеного бульйону
Хотингера
0,5
По 0,1 мл
Бактеріальна культура з
титром
109 кл/мл
0,1
3 мл
0,1 
3 мл
0,1
3 мл
0,1
3 мл
3 мл
0,7 % ПА
28
2 % ПА
Рис.1.2. Схема титрування й розсіву суспензії бактеріофага Т4В (ПА – повне агаризоване середовище)
29
Опрацювання результатів
Аналіз отриманих результатів включає в себе підрахунок кількості колоній, що виросли на чашках, і бляшок. Результати заносять у табл. 1. 2 і 1.3. Обчислюють кількість життєздатних бактеріальних клітин і частинок бактеріофага у перерахунку на 1 мл культури E. соli і суспензії фага Т4В. Крім того, варто враховувати:
•Чи є десятикратні розходження між розведеннями 10–5 і 10–6, 10–6 і 10–7 частинок/мл?
•Яка розбіжність результатів між паралельними розведеннями?
•Яка різниця між даними, отриманими двошаровим і поверхневим методами розсіву?
Таблиця 1.2
Визначення концентрації життєздатних клітин у культурі E. соli
Ряд |
Розведення |
Кількість колоній |
Концентрація |
|||
клітин у культурі |
||||||
|
|
|
|
|
||
|
|
Поверхневий спосіб розсіву |
|
|||
І |
10-5 |
|
|
|
|
|
10-6 |
|
|
|
|
||
|
10-7 |
|
|
|
|
|
ІІ |
10-5 |
|
|
|
|
|
10-6 |
|
|
|
|
||
|
10-7 |
|
|
|
|
|
ІІІ |
10-5 |
|
|
|
|
|
10-6 |
|
|
|
|
||
|
10-7 |
|
|
|
|
|
IV |
10-5 |
|
|
|
|
|
10-6 |
|
|
|
|
||
|
10-7 |
|
|
|
|
|
|
|
Двошаровий спосіб розсіву |
|
|||
І |
10-5 |
|
|
|
|
|
10-6 |
|
|
|
|
||
|
10-7 |
|
|
|
|
|
ІІ |
10-5 |
|
|
|
|
|
10-6 |
|
|
|
|
||
|
10-7 |
|
|
|
|
|
ІІІ |
10-5 |
|
|
|
|
|
10-6 |
|
|
|
|
||
|
10-7 |
|
|
|
|
|
IV |
10-5 |
|
|
|
|
|
10-6 |
|
|
|
|
||
|
10-7 |
|
|
|
|
|
30
Таблиця 1.3
Визначення концентрації життєздатних частинок бактеріофага Т4В
|
|
Кількість бляшок на чашці |
Концентрація |
|
Ряд |
Розведення |
1 |
2 |
фагових частинок, |
|
|
мл-1 |
||
І |
10-5 |
|
|
|
10-6 |
|
|
|
|
|
10-7 |
|
|
|
ІІ |
10-5 |
|
|
|
10-6 |
|
|
|
|
|
10-7 |
|
|
|
ІІІ |
10-5 |
|
|
|
10-6 |
|
|
|
|
|
10-7 |
|
|
|
IV |
10-5 |
|
|
|
10-6 |
|
|
|
|
|
10-7 |
|
|
|
Контрольні запитання
1.Охарактеризуйте морфолого-культуральні і фізіолого-біохімічні особливості E. coli.
2.Які ви знаєте типи поживних середовищ для культивування мікроорганізмів? У чому полягає роль поживних середовищ для одержання очікуваного результату при дослідженнях?
3.Що таке накопичувальна і чиста культура мікроорганізмів? Які загальні вимоги до ведення чистих культур мікроорганізмів?
4.Які ви знаєте методи визначення біомаси клітин?
5.Чим відрізняється робота з культурами в мікробіологічній і генетичній лабораторіях?
6.Назвіть основні вимоги, пропоновані для зберігання мікроорганізмів?
7.Охарактеризуйте методи нетривалого зберігання мікроорганізмів.
8.Які вам відомі методи тривалого зберігання мікроорганізмів?
9.Що означає мутантний або ревертантний вид мікроорганізмів?
10.Які принципові відмінності методів зберігання бактерій і бактеріофагів? 11.Як визначити титр бактерій? Що дає аналіз на титр? Що забезпечує успіх
аналізу?
12.Як визначити титр бактеріофага? В чому суть методу? Які істотні відмінності у визначенні титру бактерій і бактеріофагів?
Література: [1, 2, 3, 4, 12].
31
ЛАБОРАТОРНЕ ЗАНЯТТЯ 2 ЦИТОПЛАЗМАТИЧНА СПАДКОВІСТЬ У ДРІЖДЖІВ
Мета: вивчення біологічних особливостей дріжджів роду Saccharomyces; одержання клонів дріжджів і збереження їхньої генетичної чистоти.
Матеріали та обладнання: мікроскопи; термостат; центрифуга; предметні та накривні скельця; крапельниці з дистильованою водою; чашки Петрі з суслоагаром; чашки Петрі з середовищем для споруляції; камера Горяєва; стерильні піпетки і пробірки; пробірки з 2, 4,5 та 9 мл стерильної дистильованої води; культури гаплоїдного й диплоїдного штамів дріжджів Saccaromyces cerevisiae.
Загальні відомості
2.1. Дріжджі
Дріжджі, як одноклітинні гриби, є важливим біотехнологічним об’єктом і водночас зручною моделлю в дослідженні еукаріотичних організмів. Цей вибір, особливо щодо пекарських дріжджів Saccharomyces cerevisiae, пояснюється такими факторами:
•непатогенністю (для більшості видів дріжджів);
•швидким ростом;
•культура є популяцією окремих клітин, що полегшує виділення мутантів і маніпуляції з колоніями на чашках;
•існуванням стабільних гаплоїдних та диплоїдних форм, що дає можливість проводити генетичний аналіз;
•дуже активною системою гомологічної рекомбінації;
•доступністю високоефективних систем трансформації;
•малим розміром геному (1,21×107 п.н. для пекарських дріжджів), який тільки в 3,5 раза більший за геном E. coli;
•повним секвенуванням геному кількох видів дріжджів;
•розробленням нової двогібридної технології, яка дає змогу тестувати в клітинах дріжджів функціональну взаємодію певних білків різних організмів.
2.2. Характеристика будови клітин і життєвого циклу дріжджів
Вегетативний поділ клітин дріжджів зазвичай відбувається брунькуванням, під час якого дочірня клітина виникає із виросту на материнській клітині, після чого настає подвоєння ядер, утворення нової клітинної стінки і, врешті-решт, від’єднання дочірньої клітини. Розміри гаплоїдних і диплоїдних клітин змінюються залежно від фази росту і штаму. Типово, диплоїдні клітини є еліпсоїдами (5 × 6 мкм), а гаплоїди є сфероїдами діаметром 4 мкм.
Угаплоїдних клітин нові бруньки з’являються поблизу попередньої, тоді як
удиплоїдів вони виникають на протилежному полюсі клітини. Кожна материнська клітина утворює не більше 20–30 бруньок. Вік клітини можна визначити за кількістю «шрамів» на клітинній стінці, що залишаються після від’єднання дочірніх клітин.
32
Деякі диплоїдні штами S. cerevisiae можуть виявляти особливу морфологію клітин і колоній – псевдогіфи під час росту на агаризованому середовищі, дефіцитному за джерелом азоту. Ці псевдогіфові клітини є видовженими, і пари «материнська клітина – дочірня клітина» залишаються з’єднаними.
Час генерації стандартних лабораторних гаплоїдних штамів S. cerevisiae становить приблизно 90 хв на повному середовищі YPD (1% дріжджового екстракту, 2% пептону і 2% глюкози) і майже 140 хв на синтетичному середовищі в експоненційній фазі росту при оптимальній температурі 30°C. Штами зазвичай досягають найвищої густини 2×108 клітин/мл на середовищі YPD. Десятикратного титрування клітин можна досягнути лише за спеціальних умов: строгий контроль за рН середовищем, постійне додавання збалансованих поживних речовин і забезпечення ефективної аерації, що можливо лише у ферментерах.
Серед дріжджів S. cerevisiae є як гетероталічні, так і гомоталічні штами. Диплоїдні клітини обох типів можуть спорулювати за умов дефіциту поживних речовин, і особливо на спеціальному середовищі, що містить ацетат калію. Під час споруляції диплоїди входять у фазу мейозу, даючи потомство із чотирьох гаплоїдних клітин у формі спор (аскоспор), локалізованих у мішкоподібній структурі під назвою «аск». Ступінь споруляції залежить від штаму, змінюючись від кількох відсотків до майже 100%. Багато лабораторних штамів спорулюють з ефективністю вищою 50%. Більшість асків містять чотири гаплоїдні аскоспори, але бувають аски з трьома і меншою кількістю спор.
2.3. Метилотрофні дріжджі
Метилотрофні дріжджі – це еукаріотичні мікроорганізми, здатні засвоювати метиловий спирт як єдине джерело вуглецю та енергії. Серед приблизно 600 видів дріжджів, відомих на сьогодні, лише близько 50 видів здатні засвоювати метанол. Більшість із них є аскоміцетами і належать до родів: Pichia, Candida, Hansenula i
Torulopsis.
Типовими ареалами метилотрофних дріжджів є лісові, садові та заболочені ґрунти, квіти, листки, кора і сік дерев, гнилі овочі та фрукти, морські водорості, а також комахи, які живуть на деревах. Екологічна ніша характеризується наявністю пектину та лігніну – комплексних полімерів з метоксильними групами. У разі їхнього гідролізу вивільнюється метиловий спирт, який служить вуглецевим субстратом для метилотрофних дріжджів. Усі метилотрофні дріжджі є факультативнимим метилотрофами, оскільки їхній ріст можуть забезпечувати й полівуглецеві сполуки. Бродіння для метилотрофних дріжджів є нетиповим і протікає при лімітуванні кисню лише в незначній мірі. Види родів Candida i Torulopsis є аспорогенними, тоді як види метилотрофних дріжджів Hansenula і Pichia мають виражену статеву фазу, формують аски та аскоспори і піддаються техніці класичних генетичних маніпуляцій. Дисиміляційний обмін метанолу у дріжджів є відносно простий і складається з трьох послідовних ферментативних стадій окиснення метанолу до вуглекислоти.
Метилотрофні дріжджі важливі не тільки як модельні об’єкти для фундаментальних досліджень, але й як біотехнологічно цінні мікроорганізми. Вони можуть рости на дешевому субстраті, метанолі, й водночас ефективно
33
засвоювати цукри. Завдяки відсутності репресорного впливу глюкози на біосинтез дихальних ферментів у цих дріжджів, їх можна вирощувати у середовищі з високим вмістом цукру без зменшення швидкості та ефективності конверсії субстрату в біомасу. Розроблено режими культивування клітин на сумішах вуглецевих субстратів без вираженого ефекту діауксії. Дріжджі H. polymorpha є термотолерантними. Ця особливість здешевлює процедуру культивування, збільшуючи швидкість росту, полегшуючи технологію відведення тепла, перемішування (завдяки зменшенню в’язкості рідини) та спрощуючи проблему мікробіологічного забруднення. Реально існуючі та перспективні біотехнологічні процеси на основі метилотрофних дріжджів включають виробництво біомаси, ферментів, метаболітів і гетерологічних білків.
Метилотрофні дріжджі розглядають як перспективне джерело білкововітамінних концентратів, призначених для кормових і харчових цілей. У світі відчувається дефіцит білкових ресурсів, а тому отримання відносно дешевого білка одноклітинних на метанолі привертає увагу дослідників і промисловців.
Крім універсальних метаболітів (амінокислоти, цукри, нуклеотиди, вітаміни, коферменти тощо), метилотрофні дріжджі можуть служити джерелом унікальних для дріжджів продуктів – формальдегіду, мурашиної кислоти, дигідроксіацетону.
Формальдегід у промисловості синтезують шляхом каталітичного окиснення метанолу, і його світове виробництво становить мільйони тонн на рік. Здебільшого формальдегід використовують для багатотоннажного виробництва фенолта карбамід-формальдегідних смол, з яких отримують полімери. Частину цього альдегіду застосовують в тонкому органічному синтезі ліків, барвників, а також для інактивації вірусів, патогенних бактерій при виробництві вакцин.
Використовуючи мутант Candida boidinii з підвищеним рівнем синтезу алкогольоксидази, японські дослідники досягли нагромадження в середовищі інкубації з метанолом 1М формальдегіду. Аналогічно можна отримувати із етанолу оцтовий альдегід із використанням клітин дріжджів P. pastoris. Ацетальдегід – важлива сировина для синтезу багатьох органічних речовин і водночас ароматно-смакова добавка, яку використовують у харчовій промисловості для надання напоям, м’ясним, овочевим та фруктовим консервам, есенціям та жувальній гумці специфічного відчуття свіжості, а тому розроблення ефективного мікробіологічного способу отримання цієї сполуки є досить актуальною проблемою. За оптимальних умов найліпші штами метилотрофних дріжджів дають практично 100% виходу ацетальдегіду. Доведено, що клітини C. boidinii є ефективними для отримання не тільки формальдегіду, ацетальдегіду, пропіональдегіду із відповідних спиртів, а й навіть дуже токсичного акролеїну із алілового спирту.
Біотехнологічний процес конверсії метанолу в форміат розроблено з використанням іммобілізованої ферментної системи, що включає алкогольоксидазу, каталазу і бактерійну формальдегіддисмутазу, або суміші інтактних клітин дріжджів H. polymorpha та бактерій Pseudomonas putida. У цьому процесі метанол окиснюється алкогольоксидазою до формальдегіду, який формальдегіддисмутаза перетворює в форміат і метанол.
34
З огляду на широке використання у сучасній косметиці гідрофільних гелевих кремів із включеннями дигідроксіацетону, перспективним є використання метилотрофних дріжджів для мікробіологічного синтезу цього живильного для шкіри косметичного засобу. Одними із ефективних продуцентів сполуки є мутанти H. polymorpha з блоком дигідроксіацетонкінази, здатні перетворювати метанол у дигідроксіацетон. За наявності додаткового генетичного блоку гліцеролкінази такі мутанти нагромаджують у середовищі суміш тріоздигідроксіацетону і гліцерол. Описано інший метилотрофний продуцент гліцеролу – мутант C. boidinii з блоком гліцерол-3-фосфатдегідрогенази.
Метилотрофні дріжджі є перспективним джерелом для промислового виробництва амінокислот, зокрема, аланіну, глутамату, лізину, метіоніну, треоніну, валіну і триптофану. Ефективні метилотрофні дріжджі і як продуценти АТФ. Вихідним субстратом для конверсії можуть служити АМФ, аденозин або аденін. Пермеабілізовані клітини C. boidinii в разі інкубації з метанолом трансформують аденозин у АТФ до концентрації 0,2 М з виходом 77%.
Сконструйовано дуже ефективний генно-інженерний штам C. boidinii – продуцент АТФ, у якого ген аденілаткінази S. cerevisiae експресується під контролем промотора гена алкогольоксидази C. boidinii. Індуковані метанолом трансформанти мали в 10 000 разів вищий рівень активності аденілаткінази, ніж вихідні клітини, і трансформували аденозин до АТФ у 23 рази ефективніше, ніж контрольні клітини. Вихід АТФ становив 0,23 М (117 г/л) за 45 год інкубації.
У зв‘язку з високим вмістом алкогольоксидази в клітинах метилотрофних дріжджів цей флавопротеїн (як і самі клітини) можна розглядати як дешеве джерело отримання флавінаденіндинуклеотиду. Розроблено м‘який гліцероловий метод дисоціації октамерної форми алкогольоксидази із вивільненням ФАД. Крім того, ФАД можна отримувати і шляхом трансформації екзогенного рибофлавіну або ФМН клітинами метилотрофних дріжджів – до 45 мг/л. Цікаво, що ВЕРХ (HPLC)-аналіз ФАД, екстрагованого як із препаратів очищеної алкогольоксидази , так і клітин, виявив його гетерогенність і наявність, крім звичайної форми, модифікованого варіанта коферменту – т. зв. “mOФАД”.
Серед ферментів метилотрофних дріжджів найбільше практичне значення має алкогольоксидаза. Цей фермент каталізує окиснення метанолу киснем повітря з утворенням формальдегіду та пероксиду водню. Алкогольоксидаза здатна окиснювати й інші первинні аліфатичні спирти, зокрема, етанол:
СН3СН2ОН + О2 → СН3СН=О + Н2О2 На цих реакціях ґрунтується аналітичне використання алкогольоксидази для
визначення вмісту етанолу та метанолу.
За умови зниження собівартості виробництва алкогольоксидази у великих масштабах (зараз ця величина є занадто високою і оцінюється в 8 000 доларів США за 1 м лн од.), перспективним є використання ферменту в складі рідких мийних засобів, антисептичних композицій для миття посуду, ванн, біостерилізації лабораторного посуду й аналітичних пристроїв, а також у складі біостерилізуючих бинтів. Усі підходи ґрунтуються на генеруванні алкогольоксидазою (за наявності алкоголю) пероксиду водню, який відбілює тканини у першому випадку і вбиває мікроорганізми – у другому. Наприклад, при
35
внесенні у відбілюючий концентрат неіонних детергентів і 8% етанолу алкогольоксидаза стає неактивною, але реактивується при розведенні композиції
водою у 100 разів. Протягом півгодини при температурі 40°С у мийному розчині продукується до 25 мМ пероксиду водню, що цілком достатньо для відбілюючого ефекту. Слід відмітити, що використання алкогольоксидази в такій системі дало б можливість запобігти негативному впливу на довкілля широко вживаних хімічних засобів відбілювання тканин на основі перборатів.
Дріжджі як організм-господар для експресії еукаріотичних гетерологічних білків поєднують переваги молекулярно-генетичних маніпуляцій і ростових характеристик прокаріотичних організмів та субклітинного апарату посттрансляційної модифікації білків еукаріот. Порівняно із системою експресії білків у клітинах пекарських дріжджів, метилотрофні дріжджі зручніші в кількох відношеннях:
•існують добре розроблені методи отримання біомаси клітин високої густини – до 125–150 г сухої маси на літр середовища у випадку P. pastoris
і 100–130 г/л – для H. polymorpha;
•клоновано високоефективні промотори низки генів, які або регулюються метанолом (промотори генів алкогольоксидази і форміатдегідрогенази), або є конститутивними (промотор гена гліцеральдегід-3- фосфатдегідрогенази P. pastoris;
•немає гіперглікозилювання білків;
•доступність експресійних систем на основі метилотрофних дріжджів P. pastoris у формі комерційних наборів. Система експресії на основі клітин P. pastoris успішно використана для отримання більше 100 практично
важливих білків, причому продуктивність синтезу цільового продукту надзвичайно висока – до 12 г/л.
2.4. Біологічні особливості дріжджів
Дріжджі, що належать до роду Saccharomyces, становлять інтерес для генетичних досліджень, тому що є типовими еукаріотами. Водночас легко культивуються, мають високу швидкість розмноження, невелику кількість стадій у циклі розвитку і досить легко контрольовані в експерименті.
Вегетативна дріжджова клітина, як правило, диплоїдна. При утворенні аска в ній формується до чотирьох гаплоїдних спор.
Утворення диплоїда – важлива особливість життєвого циклу дріжджів, на підставі якої дріжджі підрозділяють на гомота ґетероталічні. У гетероталічних дріжджів (Saccharomyces) диплоїд утворюється в результаті копуляції гаплоїдних клітин, що належать до різних типів спарювання (позначуваних а і α). Отже, такі клітини або спори можуть копулювати тільки із клітинами й спорами комплементарного типу спарювання. Тип спарювання а або α визначається алелями одного гена.
При копуляції гаплоїдів або аскоспор різного типу спарювання утворюється гетерозиготний за цим геном диплоїд генотипу (аα) нейтральний у статевому відношенні. Якщо ізолювати одну з аскоспор будь-якого типу спарювання, то при
36
переміщенні на свіже поживне середовище вона почне розмножуватися й дасть гаплоїдний клон.
Варто відзначити, що дріжджі – ізогамні, і розходження у морфології клітин різних типів спарювання не спостерігається. Однак відомо, що гаплоїдні клітини дрібніші за диплоїдні, вони сферичної форми й при брунькуванні утворюють характерні грудки. Диплоїдні клітини овальної форми й менше комкуються при брунькуванні. Одночасно зазначені ознаки – наслідок не тільки плоїдности, а й генотипу культури.
Гомоталічні дріжджі схильні до самодиплоїдизації за рахунок злиття спор в будь-яких комбінаціях, сестриних гаплоїдних клітин або материнської клітини зі своєю брунькою.
На заняттях використовують гетероталічний штам дріжджів виду S. cerevisiae, що походить від ХІІ раси й характеризується стійкою як гаплоїдною, так і диплоїдною стадіями.
Одержання клонів дріжджів і збереження їхньої генетичної чистоти.
Вивчаючи генетику мікроорганізмів, звичайно досліджують не окремі клітини, а їхні чисті культури, під якими розуміють потомство одного виду мікроорганізмів. У разі пересівань для підтримки життєздатності чистих культур встановити зв'язок між "батьками" і "нащадками" неможливо, тому що неможливо визначити, які саме клітини розмножилися й дали цю накопичувальну культуру. Під час проведення генетичних досліджень встановлення таких зв'язків абсолютно необхідно й досягається тільки при клонуванні культури. Клон – це генетично однорідна культура, отримана в результаті безстатевого розмноження клітини, що містить одне ядро, за умови рівного розподілу генетичного матеріалу батьківської клітини, тобто мітозу.
Клон як група генетично однорідних родинних організмів є основною одиницею обліку у вивченні мінливості й спадковості мікроорганізмів. Через генетичну недовговічність клону введено поняття „штам”. Під ним розуміють клонову за походженням культуру, спадково відносно однорідну, характерні риси якої підтримуються добором за специфічними ознаками. Штам, на відміну від клону, може бути збережений під час розмноження як безстатево, так і статево. Ознаки, властиві клону, як правило зберігаються при пересіваннях і одержанні вторинних клонів (субклонів) у подібних умовах культивування. Однаковість ознак клону й субклону свідчить про стійке спадкування їх у ряді клітинних поколінь. Ряд клонових ознак використовують у генетичних дослідженнях як мітки, або маркери (наприклад, ауксотрофність, форма, розмір і колір колоній і т.д.). А також фізіологічні ознаки – стійкість до антибіотиків, отрут, випромінювання і т.д.
Знання методів клонування необхідно не тільки для проведення генетичних досліджень, а й для вирішення практичних завдань – збереження й підтримки певних властивостей колекційних штамів, мікроорганізмів. Особливо важливо для збереження виробничо-коштовних штамів – попередити генетичне виродження штамів у результаті зберігання й частих пересівань. Із цією метою слід не пересівати на свіже поживне середовище, а здійснювати розсів цих штамів для

37
відбору клонів, що мають батьківські властивості культур, і для вибракування спонтанних мутантів.
Завдання на виконання
1.Для одержання окремих клонів методом розсіву моноклітинної суспензії готують суспензію, для чого в чашку Петрі з пятидобовою культурою дріжджів (засіяною суцільним газоном) додають 7 мл дистильованої води. Культуру змивають шпателем. Суспензію дріжджів піпеткою місткістю 10 мл переносять у склянки для центрифугування.
2.Після зрівноважування склянок суспензію центрифугують протягом 5 хв при обертовій частоті 1000 об/хв. З надосадової рідини спеціально підготовленою піпеткою місткістю 10 мл відбирають 3 мл і переносять у пробірку.
3.Відбирають дві петлі суспензії й переносять у камеру Горяєва. Підраховують кількість клітин і визначають концентрацію культури за формулою:
A = 20n 106 ,
де n – сума клітин у п’яти великих квадратах.
4.Прийнятний режим центрифугування, коли у вихідній суспензії має міститися приблизно 106 клітин/мл. Отриману в пробірці суспензію розводять приблизно в 100 разів. Кількість колоній після висіву не повинна перевищувати
500.
5.Для розведення беруть дві пробірки з 4,5 мл стерильної дистильованої води. У першу вносять 0,5 мл суспензії, потім з неї переносять 0,5 мл у другу. Із другої проводять розсів на щільне поживне середовище. У попередньо марковані чашки заливають поживне середовище. Після його застигання на поверхню наносять 0,05 мл суспензії. Потім стерильним шпателем розтирають суспензію по поверхні середовища. Чашки вміщують у термостат з температурою 30 °С. Схема проведення досліду зображена на рис. 2.1.
6.Переглянути під мікроскопом (збільшення 400 разів): вегетативні клітини гаплоїдного й диплоїдного штамів дріжджів; препарати „роздавлена крапля” у дистильованій воді. Визначити форму й розмір клітин (замалювати);
7.Висіяти диплоїдні й гаплоїдні штами на поживне середовище для
споруляції; петлею зробити густий висів на поверхню середовища (приблизно 1 см2). Вирослу культуру розглядають на наступному занятті;
8.Приготувати у краплині води препарати з культур дріжджів з різною плоїдією; промікроскопіювати диплоїдні клітини, підрахувати кількість вегетативних клітин і асків у кількох полях зору, вивести відсоток споруляції; промікроскопіювати гаплоїдні культури, переконатися у відсутності споруляції; результати занести в табл. 2.1.

38
7 мл
ПА Газон
Стерильна
Суспензія
Центрифугуванн |
||
|
я, |
|
1 петля |
3 мл надосадової |
|
|
2 петлі |
|
2 мл |
|
|
стерильної |
|
|
води |
|
|
1 петля |
Камера Горяєва, |
|
0,5 мл |
||
кількість, монотиповість |
||
|
4,5 мл |
|
|
ПА, |
стерильної |
|
|
води |
|
||
Висів |
0,05 мл |
||
|
|||
штрихом |
|
||
|
|
ПА,
Титр
Рис.2.1. Схема виконання дослідів і виділення клонів (ПА – повне агаризоване середовище)
39
Таблиця 2.1
Морфологічні особливості диплоїдних і гаплоїдних культур дріжджів
№ |
Клітини |
Тип |
Кількість |
|
Споруляція, |
||
|
|
розташування в |
|
|
|
||
|
|
Вегетативних |
|
|
|||
п/п |
Форма |
Розмір |
|
Асків |
% |
||
мазку |
клітин |
|
|||||
|
|
|
|
|
|
||
1 |
|
|
|
|
|
|
|
2 |
|
|
|
|
|
|
|
3 |
|
|
|
|
|
|
|
4 |
|
|
|
|
|
|
|
5 |
|
|
|
|
|
|
|
6 |
|
|
|
|
|
|
|
9. Для одержання окремих колоній виснажуючим штрихом у пробірку з 2 мл стерильні води вносять одну петлю дріжджової культури, ретельно перемішують. На твердому середовищі петлею проводять горизонтальний штрих суспензією з пробірки. Петлю обпалюють, охолоджують, проводять нею по поверхні середовища кілька вертикальних штрихів і знову обпалюють. Далі петлю охолоджують і проводять по поверхні середовища горизонтальні штрихи, що перетинають вертикальні, не торкаючи перший горизонтальний штрих. Розсів проводять на двох чашках.
Опрацювання результатів
Аналізи виконують після чотирьохдобового вирощування дріжджів у термостаті за температури 30°С. Визначити кількість вирослих колоній, визначити титр клітин шляхом підрахунку у камері Горяєва і методом висіву на агаризоване середовище, дослідити монотиповість і гетерогенність вирослих колоній візуально й за допомогою бінокулярної лупи (мікроскопа МБС-1). Результати занести у табл. 2.2.
|
|
|
|
|
|
|
Таблиця 2.2 |
|
|
Аналіз диплоїдних і гаплоїдних культур дріжджів |
|||||
|
|
|
|
|
|
|
|
№ |
Штам |
|
Характер |
Титр клітин |
Кількість колоній |
||
|
Камера |
|
|
|
|||
п/п |
|
аналізу |
Висів |
монотипових |
гетеротипових |
||
|
|
Горяєва |
|||||
1 |
|
|
|
|
|
|
|
2 |
|
|
|
|
|
|
|
3 |
|
|
|
|
|
|
|
4 |
|
|
|
|
|
|
|
5 |
|
|
|
|
|
|
|
6 |
|
|
|
|
|
|
|
40
Контрольні запитання
1.Чому дріжджі Saccharomyces cerevisiae є важливим об’єктом біотехнологічних досліджень?
2.Охарактеризуйте морфологічні особливості гаплоїдних і диплоїдних клітин дріжджів.
3.Дайте характеристику метилотрофним дріжджам.
4.Яке практичне значення метилотрофних дріжджів?
5.Які переваги використання метилотрофних дріжджів у порівнянні з хлібопекарськими?
6.Які генетичні відмінності дріжджів і бактерій? У чому їх спільність?
7.Що означає поняття "гаплоїдна", "диплоїдна", "поліплоїдна" клітини?
8.Що означають поняття "штам", "клон", "асоціація"? Які їх спільні і відмінні ознаки?
9.В чому полягає суть методу клонування мікроорганізмів? Як його використовують у практичних цілях?
Література: [1, 2 6, 8,9].
ЛАБОРАТОРНЕ ЗАНЯТТЯ 3 ОДЕРЖАННЯ ГІБРИДІВ І ПРИНЦИПИ ГІБРИДОЛОГІЧНОГО
АНАЛІЗУ Мета: ознайомлення з методами схрещування клітин мікроорганізмів;
виділення гібридів мікроорганізмів; одержання гібридних аскоспор і розсів отриманих аскоспор на чашки.
Обладнання та матеріали: термостат; чашки Петрі з мінімальним (М); повним (П); ацетатним середовищами (А); пробірки з 0,5 мл травного соку равликів (ТСР); 1 пробірка з 1 мл стерильної води; камера Горяєва; шпателя; стерильні піпетки; колба зі стерильною дистильованою водою; два штами дріжджів S. cerevisiae.
Загальні відомості 3.1. Схрещування клітин мікроорганізмів
На основі гібридизації мікроорганізмів можна, по-перше, з'ясувати характер успадкування тієї або іншої ознаки, що визначається ядром або цитоплазмою й залежить від одного або кількох генів і, по-друге, одержати гібриди з цінними для використання у виробництві батьківськими ознаками.
Методи одержання гібридів і вивчення розщеплення в їхньому потомстві визначається особливостями життєвого циклу досліджуваного організму. Гібридизація може здійснюватися як у гетероталічних, так і в гомоталічних організмів.
Для розшифрування структури генетичного апарата клітини аналізують розщеплення в потомстві гібридів за чітко наслідуваними ознаками (маркерами). Для виділення гібридних клітин використовують мікроманіпулятор або проводять
41
висів на спеціальні середовища, на яких селекціонують колонії гібридних клітин (за культуральними ознаками).
Якщо ознаку зумовлює один ген, у потомстві спостерігють розщеплення у відношенні 1:1 (АВ), що випливає із закономірної розбіжності гомологічних хромосом у мейозі, разом з якими розподіляються й алелі розглянутого гена. Якщо реєструють розщеплення за двома ознаками, причому кожна визначається одним геном і обоє вони розташовуються на різних парах хромосом, тобто незалежно комбінуються, то спостерігають розщеплення у відношенні 1:1:1:1 (якщо алелі одного гена позначити А і а, а іншого В і в), генотипи аналізованого гібрида Аавв і при мейозі 25 % аскоспор будуть мати генотип АВ, 25 % – Ав, 25 % – аВ і 25 % – ав. Якщо розщеплення за однією ознакою буде відрізнятися від АВ, це свідчить про те, що досліджувану ознаку визначають не один, а кілька генів.
3.2. Виділення гібридів мікроорганізмів
Для виконання роботи використовують культури аденинзалежних мутантів дріжджів S. cerevisiae (вони несуть неалельні мутації аденинзалежності ade1 і аdе2), які мають забарвлені колонії (ефект тих самих мутацій): штам S. cerevisiae a ade2 ADE1 ALC4 – червоні, а штам α ADE2 ade1 alc4 – руді. Мутація аlс4, головним проявом якої є дихальна недостатність, змінює забарвлення колонії з червоного на рудий.
Гібрид, отриманий від схрещування цих штамів, може бути відібраний на мінімальному середовищі без аденіну. Він буде мати генотип aα ADE1 ade1 ADE2 ade2 ALC4 alc4.
Мутації ade1 і ade2 є комплементарними одна одній.
Одержання гібридних аскоспор і розсів отриманих аскоспор на чашки.
Аскоспори, утворені таким гібридом, мають такі генотипи:
a |
ade1 |
ade2 |
alc4 |
α |
ade1 |
Ade2 |
alc4 |
a |
ade1 |
ade2 |
ALC4 |
α |
ade1 |
Ade2 |
ALC4 |
a |
ade1 |
ADE2 |
alc4 |
α |
ade1 |
ADE2 |
alc4 |
a |
ade1 |
ADE2 |
ALC4 |
α |
ade1 |
ADE2 |
ALC4 |
a |
ade1 |
ade2 |
alc4 |
α |
ADE1 |
Ade2 |
alc4 |
a |
ADE1 |
ade2 |
ALC4 |
α |
ADE1 |
Ade2 |
ALC4 |
a |
ADE1 |
ADE2 |
alc4 |
α |
ADE1 |
ADE2 |
alc4 |
a |
ADE1 |
ADE2 |
ALC4 |
α |
ADE1 |
ADE2 |
ALC4 |
Примітка. До риски розміщено генотипи аскоспор, що утворюють забарвлені колонії, нижче риски – білі, як і вихідний гібрид.
Після розсіву суспензії аскоспор можна врахувати розщеплення за мутацією аlс4, що проявляється в модифікації забарвлення, тобто врахувати генотипи ALC4 і alc4. Серед забарвлених колоній співвідношення червоних і рудих очікується 1:1, тобто таке ж, як спостерігалося б серед усього аскоcпорового потомства, якби ми врахували генотипи ALC4 і аlс4 не тільки серед забарвлених, а й серед безбарвних колоній.
42
Завдання на виконання
1. Готують суспензію взятих для дослідження двох штамів дріжджів S. cerevisiae, для чого вносять петлю кожної культури в пробірку з 1 мл стерильної води. Для приготування суспензії дріжджовий матеріал слід брати з типово забарвлених колоній розсіву (на чашках).
2.Стерильні чашки заливають середовищами М та П і маркують: на дні кожної чашки по склу проводять три паралельні лінії. На крайніх лініях наносять позначення батьківських штамів. Середню лінію позначають маркером, який вказує на спільний висів обох батьківських культур. Після маркування чашок із пробірок петлею відбирають частину приготовленої суспензії й висівають по крайньому й середньому штрихом, потім іншу суспензію висівають по іншому крайньому штриху й середньому штриху поверх іншої культури. Засіяні чашки вміщують у переверненому виді в термостат при температурі 30 ± 1 °С. Період вирощування – 7 діб.
3.Проводять аналіз вирослої культури за штрихами на двох середовищах: із середнього штриха вирослої гібридної культури на середовищі М петлею проводять пересівання дріжджів на чашки із середовищем П. Висів слід робити виснажуючим штрихом. Чашки в переверненому вигляді поміщають у термостат при температурі 30 ± 1 °С на чотири доби.
4.Розглядають ріст гібрида на чашках. Переносять клітини гібриду на середовище з ацетатом, беручи їх з окремих великих білих колоній. Матеріал (стільки, скільки утримається на краю петлі) розподіляють по поверхні
середовища з ацетатом на площі 5 × 10 мм. Чашки із висівом дріжджових гібридів вміщують у термостат з температурою 30 ± 1 °С на 2 – 3 доби.
Опрацювання результатів
1.З вирослої на зазначеній площі ацетатного середовища культури петлею збирають клітини гібриду й переносять у пробірку з 0,5 мл травного соку равликів. Культуру збовтують і залишають у спокої на 2 – 3 год при кімнатній температурі. Після закінченні цього часу у пробірку додають 4 мл стерильної дистильовані води. Суспензію ретельно збовтують.
2.Для одержання суспензії з концентрацією дріжджових клітин 5000 і 1000 клітин/мл проводять розведення, для чого на початку визначають вміст спор у суспензії підрахунком їх у камері Горяєва, а потім розводять до потрібної концентрації, додаючи 0,5 мл суспензії до 4,5 мл стерильної води. Із двох останніх розведень проводять висів на чашки із середовищем П, наносячи на поверхню 0,1 мл суспензії з наступним розтиранням її шпателем. Засіяні чашки в переверненому вигляді вміщують у термостат з температурою 30 ± 1 °С на сім діб.
3.Для гібридологічного аналізу на чашках підраховують кількість колоній червоного і рудого кольору (оптимальна густина висіву 200 колоній на чашку).
Отримані результати обробляють за критерієм χ2 (ксі-квадрат) за допомогою формули: