

Процесс носит название «деалкилирующего нитрования»
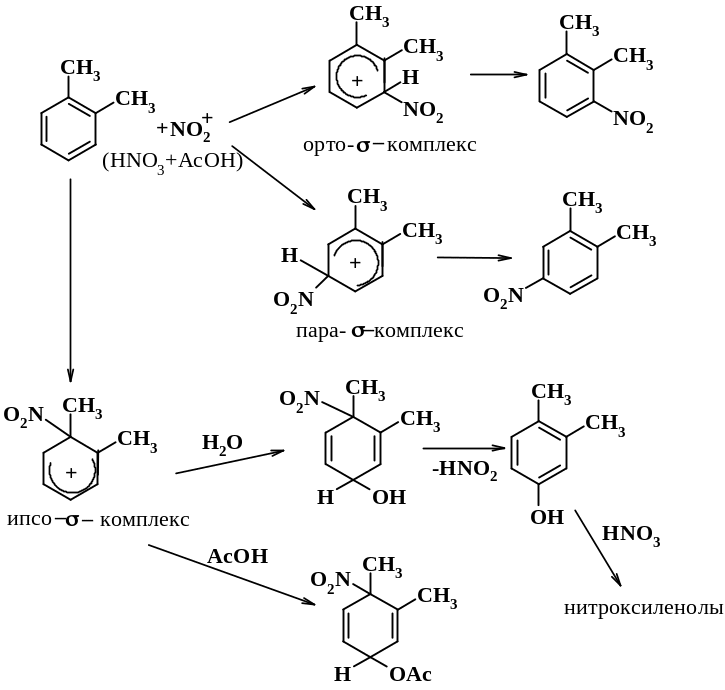
Схема 9.1.7 - Нитрование орто-ксилола
Таким образом, пути стабилизации ипсо-σ-комплексов таковы:
1. захват нуклеофила; 2. перегруппировка; 3. 1,2-(1,3)-миграция заместителя X; 4. потеря заместителя (ипсо-замещение); 5. потеря протона или родственной группы от заместителя, удаленного от ипсо-положения;
6. возврат к π-комплексу или исходному продукту.
9.2 Ориентация как отражение свойств σ-комплекса
Ориентацию электрофильного замещения в бензольном кольце можно объяснить как результат электронодонорного или - акцепторного эффектов заместителей в исходном (статическом) состоянии молекулы арена, так и с точки зрения стабильности промежуточного продукта - σ-комплекса (аренониевого иона) (динамический эффект).
При этом полагают, что переходное состояние (ПС) медленной стадии нитрования по структуре близко к аренониевому иону, хотя это не всегда справедливо (см. стр. 50). Заместитель Х в молекуле C6H5X может стабилизировать или дестабилизировать этот карбокатион, поэтому позиционная селективность должна соответствовать образованию наиболее стабильного из изомерных σ-комплексов.
Рассмотрим влияние сильных активирующих заместителей для орто-, мета- и пара-замещения в анизоле. Для орто-замещения в анизоле две граничные структуры аренониевого иона представляют собой вторичный карбокатион, а третья - третичный, где положительный заряд дополнительно стабилизирован за счет участия неподеленной пары электронов атома кислорода группы OCH3 с образованием оксониевой структуры (+М-эффект) (схема 9.2.1):

Схема 9.2.1
Для пара-замещения в анизоле две предельные структуры также являются вторичными карбокатионами, а третья – третичным, дополнительно стабилизированным соседней метоксигруппой (схема 9.2.2)
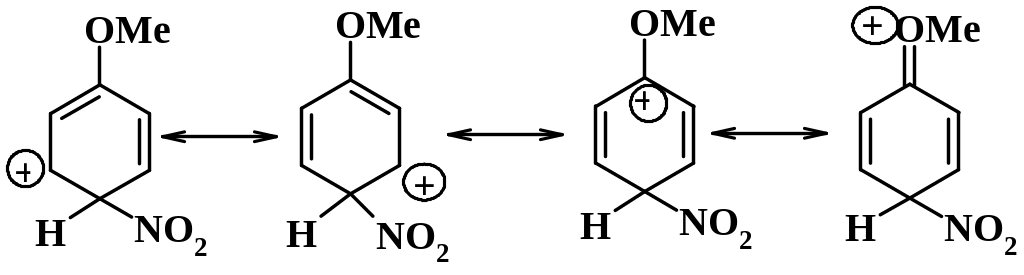
Схема 9.2.2
Стабилизация циклогексадиенильного катиона за счет +М-эффекта метокси-группы облегчает замещение в орто- и пара-положениях анизола по сравнению с бензолом и ориентирует входящую группу NO2 именно в эти два положения ядра. Для мета-замещения такая стабилизация невозможна,
т ак
как ни в одной из предельных структур
положительный заряд не может
находиться у атома углерода, несущего
метокси-группу (схема 9.2.3). Кроме того,
мета-аренониевый ион анизола
дестабилизирован –I-эффектом
группы OCH3:
ак
как ни в одной из предельных структур
положительный заряд не может
находиться у атома углерода, несущего
метокси-группу (схема 9.2.3). Кроме того,
мета-аренониевый ион анизола
дестабилизирован –I-эффектом
группы OCH3:
Схема 9.2.3
Аналогичная картина наблюдается при замещении в феноле, анилине и других аренах, содержащих +М-заместители. В толуоле и других алкилбензолах стабилизация третичного карбокатиона достигается за счет гиперконьюгации с метильной или другими алкильными группами (см. раздел 3, стр.22). Поэтому ПС в толуоле при орто- и пара–замещении обладает меньшей энергией по сравнению с бензолом. Для мета-замещения в толуоле все три предельные структуры σ-комплекса соответствуют вторичным карбокатионам, слабо стабилизированным лишь за счет +I- эффекта группы CH3 (схема 9.2.4).

Схема 9.2.4
Поэтому мета-положение в толуоле более активно, чем при атаке ионом нитрония молекулы бензола, но менее активно, чем орто- и пара-положения.
![]() Для галогенбензолов ситуация отличаемся
от вышеприведенных примеров ввиду
сильного –I-эффекта
галогена. При замещении в мета-положение
карбокатионы, соответствующие
двум последним граничным формулам
(схема 9.2.5), сильно дестабилизированы
электростатическим взаимодействием
карбокатионного центра с положительным
концом диполя (структуры b
и с).
Для галогенбензолов ситуация отличаемся
от вышеприведенных примеров ввиду
сильного –I-эффекта
галогена. При замещении в мета-положение
карбокатионы, соответствующие
двум последним граничным формулам
(схема 9.2.5), сильно дестабилизированы
электростатическим взаимодействием
карбокатионного центра с положительным
концом диполя (структуры b
и с).


Схема 9.2.5 Схема 9.2.6
Однако для замещения в орто- и пара-положениях (схема 9.2.6) предельные структуры также дестабилизированы электростатическим взаимодействием диполя с карбокатионным центром по сравнению с бензолом; тем не менее, в одной из структур (а) возможна стабилизация за счет +М- эффекта галогена. Галогенониевые ионы менее стабильны, чем иммониевые или оксониевые, поэтому орто-пара-ориентирующий эффект галогена наблюдается на фоне общей дезактивации арена.
![]() Влияние мета-ориентирующих
заместителей X (X
= NO2, CN,
COR и др.) сводится к сильному
электростатическому отталкиванию между
карбокатионным центром и положительным
концом диполя X (например,
).
Влияние мета-ориентирующих
заместителей X (X
= NO2, CN,
COR и др.) сводится к сильному
электростатическому отталкиванию между
карбокатионным центром и положительным
концом диполя X (например,
).
Поэтому все положения ядра
дестабилизированы по сравнению с
бензолом, но наиболее сильная дезактивация
наблюдается в орто- и
пара-положениях, где в одной из
предельных структур положительно
заряжены соседние атомы C
и N (схема 9.2.7, а) и b)),
а в случае мета-замещения (схема
9.2.7, с)) два положительных заряда разделены
одним или тремя а томами
углерода цикла.
томами
углерода цикла.
Схема 9.2.7
Поэтому мета-замещение здесь дезактивировано в меньшей степени, чем орто- и пара-, и преобладает мета-изомер.